Курсовая работа: Влияние антигололедного покрытия на коррозионную стойкость углеродистой стали
МОРДОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
ИМЕНИ Н.П. ОГАРЕВА
Институт физики и химии
Кафедра аналитической химии
КУРСОВАЯ РАБОТАВЛИЯНИЕ АНТИГОЛОЛЕДНОГО ПОКРЫТИЯ НА КОРРОЗИОННУЮ СТОЙКОСТЬ УГЛЕРОДИСТОЙ СТАЛИ
Автор курсовой работы 27.06.2011 М.В. Девяткина Обозначение курсовой работы КР-02069964-020101-04-11
Специальность 020101 химия
Руководитель работы
канд. хим. наук, доцент 27.06.2011 А.К. Осипов
Оценка
Саранск 2011
МОРДОВСКИЙ ГОСУДАРСТВЕННЫЙ УНИВЕРСИТЕТ
ИМЕНИ Н.П. ОГАРЕВА
Институт физики и химии
Кафедра аналитической химии
ЗАДАНИЕ НА КУРСОВУЮ РАБОТУСтудентка Девяткина М.В.
1 Тема: Влияние антигололедного покрытия на коррозионную стойкость углеродистой стали
2 Срок предоставления работы к защите: 27.06.10 г.
3 Исходные данные для курсовой работы: литературные данные, постановка цели и задачи исследования
4 Содержание курсовой работы
4.1 Аналитический обзор
4.2 Экспериментальная часть
4.3 Результаты и их обсуждение
4.4 Выводы
Руководитель курсовой работы 8.02.2011 Осипов А.К.
Задание принял к исполнению 8.02.2011 Девяткина М.В.
Содержание
Введение 4
1 Аналитический обзор 6
1.1 Коррозионное поведение углеродистых сталей в растворах солей 6
1.2 Анодное поведение углеродистых сталей и цветных металлов в органических растворах 9
1.2.1 Азотсодержащие соединения 10
1.2.2 Соединения с кратными связями 14
1.2.3 Кислородсодержащие соединения 17
1.3 Растворение и пассивация железа в щелочных растворах 19
2 Экспериментальная часть 22
2.1 Электрохимические методы анализ 22
2.2Методика потенциометрического определения 24
2.3 Подготовка электродов для электрохимических измерений 26
2.4 Приготовление рабочих растворов 28
3 Результаты и их обсуждение 29
3.1 Анодное поведение углеродистой стали в исходных растворах 29
3.2 Анодное поведение стали Ст 85 в смесях солей 31
3.3 Скорость коррозии в чистых растворах 34
4 Выводы 36
5 Список использованных источников 37
Введение
В зимний период времени возникают не малые трудности в эксплуатации транспортных магистралей, прежде всего городских дорог, бетонных дорожек аэропортов, аэродромов в силу образования гололеда.Главной задачей уборки магистралей и улиц в зимний период является предупреждение образования наледи на дорогах. Основной технологической операцией зимней механизированной уборки является обработка проезжей части противогололедными материалами.
Зачастую дорожные службы, эксплуатационники не учитывают степень агрессивного воздействия применяемых антигололедных реагентов на транспортные средства и покрытия, особенно этим страдают коммунальные службы населенных пунктов, где основным средством борьбы с гололедом является галит (каменная соль). Завоевавшим наибольшую популярность в борьбе с гололедом, а также наиболее дешевым средством является галит (соль техническая, хлорид натрия). Также используется смесь галита (соли технической, натрия хлористого, хлорида натрия) с песком и щебнем, которые добавляются для уменьшения коэффициента скольжения и увеличения сцепных качеств (пескосоляные смеси).Лишь на более важных объектах прибегают к использованию менее агрессивных средств, но опять же не учитывая экономическую сторону вопроса.
В зависимости от климатических условий и интенсивности движения автомобилей в зимние периоды года при гололеде ежедневно расходуется от 10 до 150 г соли (NaCl) на 1 м2 дорожного покрытия.
Темой нашего исследования явилось снижение агрессивного влияния антигололедного реагента путем введения веществ, снижающих коррозионные потери конструкционных материалов машин и дорожных покрытий.
Многие из факторов, характеризующих химические и физические свойства среды окружающей систему, оказывают глубокое влияние на природу и ее интенсивность. Тривиально, но очевидный факт, свидетельствует о снижении коррозии с падением кислотности раствора. Когда раствор становится щелочным и коррозия уменьшается, наступает естественное ингибирование.
Поэтому, целью настоящей курсовой работы является исследование влияния антигололедного покрытия на коррозионную стойкость углеродистой стали Ст.85.
Задачи курсовой работы:
1) провести коррозионные испытания стали Ст.85 в растворе галита.
2) провести электрохимические испытания стали Ст.85 в растворах галита с добавлением различных солей.
3) провести электрохимические испытания образца арматуры Ст.85, в растворе тройной системы “галит-мочевина-гидрокси́д ка́льция” в разных соотношениях.
1 Аналитический обзор
1.1 Коррозионное поведение углеродистых сталей в растворах солей
С.М. Решетников отмечает участие анионов фона, в частности ОН- -ионов, в промежуточных стадиях процесса. Адсорбция ПАВ влияет на соотношение скоростей стадий анодного процесса и его кинетические параметры [1, 2—5].На примере анодного растворения железа в сульфатных растворах рассмотрим влияние ингибиторов.
Схема анодного процесса в присутствии ингибиторов*:
Fe + H2 0 + Inh ↔[Fe(OH)(Inh)]адс + Н+ + е- (3.1а)
[Fe(OH) (Inh)]адс + S02-4 ↔[Fe(OH)(SO4 ) ]2-адс +Inh (3.1б)
[Fe(OH)(SO4 ) ]2-адс →Fe2+ + ОН- +S02-4 + е- (3.1в)
С.М. Решетниковым рассматривается как одновременно протекаю-
щая адсорбция молекул воды, которая завершается образованием
ОН-адс и адсорбция частиц ПАВ. Будет ли это сопровождаться ингибированием или стимулированием анодного процесса, зависит от прочности адсорбционного комплекса, образовавшегося на этой стадии, его способности претерпевать дальнейшие превращения в конечный продукт анодного процесса — гидрйтированные ионы Fe+2 .
Стадию (3.1б) рассматривается как конкурирующую адсорб-
ция анионов сульфата, имеющихся в растворе, и частиц ПАВ. Эта ста-
дия завершается вытеснением из адсорбционного комплекса частиц ин-
гибитора с образованием соединения, в состав которого входят два
вида анионов, ускоряющих процессанодного растворения.
Конечный продукт анодного растворения железа—ионы Fe+2 —появляется в ходе стадии (3.1в), которая определяет скорость всего процесса.
С.М. Решетников используя преобразования, получает следующее кинетическое уравнение:
jа = kа [OH-]°,5p [Inh]0,5р-1 exp [(p + 1) FE/RT](3.2)
При р < 2 величина vinh <0 и ПАВ будет уменьшать скорость анодной реакции, т. е. служить ингибитором. При р > 2 величина vinh > 0 и ПАВ будет увеличивать скорость анодной реакции, т. е. служить стимулятором. Иначе говоря, в зависимости от отношения
степеней заполнения (р) поверхности адсорбционными комплексами, содержащими и не содержащими ПАВ (а это, в свою очередь, определяется в частности прочностью адсорбционных связей ПАВ — металл), будут реализоваться случаи ингибирования (р<2), стимулирования (р>2)или независимости (р=2) скорости анодного процесса от наличия ПАВ.
С учетом поверхностной активности ионов сульфата на железе
обосновывается другая схема анодного процесса:
![]() Fe +S02-4 + Inh ↔[Fe(SO4 )(Inh)]-адс + е- (3.4a)
Fe +S02-4 + Inh ↔[Fe(SO4 )(Inh)]-адс + е- (3.4a)
[Fe(SO4 )(Inh)]-адс + H2 O ↔[Fe(SO4 )(OH)]2-адс + H+ + Inh (3.46)
[Fe(SO4 )(OH)]2-адс → Fe2+ + SO2-4 + OH- + е- (3.4b)
Схема 3.4 предусматривает одновременную адсорбцию ионов сульфата и ингибитора на стадии (3.4а) с последующим адсорбционным
вытеснением ингибитора водой на стадии (3.46).Говорится о приблизи-
тельно одинаковой адсорбционной активности ионов сульфата и гидр-
оксида в данных условиях. Распространяя это на ингибированные рас-
творы, можно полагать, что схемы (3.1) и (3.4) равновероятны и про-
текают пераллельно, тем более, что кинетически они неразличимы.
Анодное растворение железа в кислых хлоридных растворах имеет
ряд характерных особенностей, что связано с участием в процессе анионов хлорида.Схема анодного процесса в присутствии ингибиторов:
![]() Fe + H2 O + Inh ↔[Fe(OH)(Inh)]адс + H+ + е-
Fe + H2 O + Inh ↔[Fe(OH)(Inh)]адс + H+ + е-
[Fe(OH)(Inh)]адс + Cl- ↔[Fe(OH)(Cl)]-адс + Inh (3.5)
[Fe(OH)(Cl)]-адс → Fe2+ + OH- + Cl- + е-
Этой схеме соответствует уравнение
jа = ka [ОН- ]0.5p [Inh]0.5p-1 exp[(p + 1) FE/2RT](3.6)
Из всех ионов наиболее агрессивным, по литературным данным, является ион хлора, который в силу этих причин был объектом многочисленных исследований.Агрессивность иона хлора объясняют различным образом. Так, например, Томашов и Модестова [6] указывают, что галоидный анион разрушает окисную защитную пленку, причем скорость разрушения окисных пленок возрастает с увеличением концентрация ионов хлора. Ванюкова и Кабанов [7] считают, что агрессивность иона хлора обусловлена способностью его к адсорбционному вытеснению кислорода с поверхности металла. Акимов [8] агрессивное действие ионов объясняет тем, что ионы способны проникать через пленку, защищающую металл от коррозионной среды. Хедже [9] устанавливает ряд анионов, расположенных в порядке уменьшения их проникающей силы:
Cl — > Br- > J- > F- > SO2-4 > NO-3 > PO3-4
Из этого ряда видно, что активирующая сила анионов, в основном, уменьшается с увеличением размеров ионов.
Н.Г. Чен при обработке опытных данных делает вывод, что хлорид исульфат натрия при одинаковых условиях иконцентрациях 400 мг/л разрушают металл различно: раствор Na2 SO4 оказывается более агрессивным. Однако активирующее действие сульфата падает, примерно, в 1.5 раза, если раствор содержит50мг/л фосфатного замедлителя коррозии.Приприбавлении к этому раствору 400 мг/л хлорида натрия коррозия увеличивается в 1,2раза. Если добавить NаС1 в 2раза меньше, т.е.200мг/л, токоррозия возрастает только в 1,1раза. Таким образом, хлорид натрия, содержащийся в агрессивной среде, увеличивает коррозию металла, причем тем больше, чем выше его концентрация в растворе.Исследование коррозии металла в кипящих растворах NaCl, Na2 SO4, СаСl2 и CaS04 показывает, что скорость коррозии с возрастанием концентрации их растворов во всех случаях увели-
чивается, причем у сульфата натрия имеется область концентраций (100—1000 мг/л), при которой наблюдается повышенная скорость коррозии. Если эти соли расположить в порядке уменьшения их агрессивности, то получим следующие ряды:
Na2 SO4 > NaCl > СаС12 > CaS04
и при более высокой концентрации, растворов:
NaCl > СаСl2 > Na2 S04 > CaS04
При рассмотрении этих рядов обращают на себя внимание два об-
стоятельства. Во-первых, соли Na2 SO4 и NaCl имеют одинаковые катио-
ны Na+, но различные анионы S042- и С1-. Следовательно, различное их
агрессивное действие обусловлено только различием их анионов. При
концентрациях раствора ниже 1000 мг/л анион S042- является более аг-
рессивным, чем ион С1-. Во-вторых, соединения Na2 S04 и CaS04, NaCl и
СаС12 имеют соответственно одинаковые анионы, но катионы у них раз-
личные. Это весьма сильно сказывается на их агрессивных свойствах:
при наличии в растворе ионов Са2+ сильно снижается агрессивное дей-
ствие анионов, особенно SO2-4 .
1.2 Анодное поведение углеродистых сталей и цветных металлов в органических растворах
Защита металлов от коррозии ингибиторами, как было пока-
зано, часто связана с химической адсорбцией, включающей изме-
нение заряда адсорбирующегося вещества и перенос заряда с од-
ной фазы на другую. Поэтому особое значение приобретает мо-
лекулярная структура ингибиторов. Электронная плотность на
атомах функциональных групп, являющихся реакционным цент-
ром, влияет па прочность абсорбционной связи. Кроме того, проч-
ность связи зависит и от свойств металла, а также поляризуемо-
сти функциональной группы.
В состав большей части органических ингибиторов входит по
крайней мере одна полярная группа с атомом азота, серы, кисло-
рода, а в некоторых случаях селена и фосфора.
В связи с этим большой интерес представляют работы, в ко-
торых пытаются связать ингибирующие свойства органических со-
единений с их структурными особенностями. В этой области об-
ширные исследования были проведены Хаккерманом [10], кото-
рый сформулировал основные положения адсорбционной теории
органических ингибиторов. По этой теории ингибирующие свой-
ства многих соединений определяются электронной плотностью на
атоме, являющемся основным реакционным центром. С увеличе-
нием электронной плотности у реакционного центра хемосорбци-
онные связи между ингибитором и металлом усиливаются. Иссле-
дуя ингибирующие свойства пиридина и его производных, Хак-
керман установил, что защитные свойства этих соединений, т. е.
способность уменьшать коррозию, действительно увеличиваются
по мере увеличения электронной плотности на атоме азота в ряду:
пиридин < 3-пиколин< 2-пиколин< 4-пиколин.
То же наблюдается у алифатических и циклических аминов:
чем больше электронная плотность на атоме азота, тем более эф-
фективен ингибитор. Циклические амины — лучшие ингибиторы по сравнению с алифатическими аминами, у которых электронная
плотность на азоте существенно меньше.
Аналогичное исследование ингибирующих свойств ароматических аминов и тиолов и их производных было выполнено Риггосом [11], которыйвсогласиистеориейполучилхорошеесовпадение
экспериментальныхрезультатовстеоретическимипредпосылками,
связывающимизащитныесвойстваингибиторовсэлектронной
структурой.
Трабанелли, ФисупаиКарасситти[12]распространилиэтивоз-
зрениянамеханизмдействиялетучихингибиторовкоррозии.Изу-
чаяалициклическиеамины(циклогексиламинидициклогексил-
амин)иихпроизводные, авторыустановили, чтоэффективность
нгибиторовзаметнорастетсувеличениемэлектроннойплотности
наатомеазота, котораяопределяетсяэлектронно-отталкивающим
эффектомзаместителей.
Всвязисэтимособыйинтереспредставляетопределениеэлек-
троннойплотностинаатоме, являющимсяцентромреакции, атак-
жеустановлениеосновныхзакономерностейееизменения.Особое
значениепоследнееимеетдляароматическихигетероциклических
соединений, посколькуунихэлектроннаяплотностьнареакцион-
номцентреможетсильноменятьсяпривведенииразличныхза-
местителей.
Эффективностьфункциональногоатомавадсорбционныхпроцессахприравнойстабильностисоединенийизменяетсявследующемряду: селен>сера>азот>кислород, что, помнениюТрабанеллииКарасситти[12], можетбытьобъясненоменьшейэлектроотрицательностьюэлементовслева, вследствиечегоихсоединениялегчеполяризуются.
1.2.1 Азотсодержащие соединения
Азотсодержащие соединения, проявляющие свойства ингибиторов кислотной коррозии, — это алифатические, ароматические, нафтеновые и гетероциклические амины, амиды, хинолины и др. Значительная часть промышленных ингибиторов-органические азотсодержащие ПАВ. Наиболее исследованы в виде индивидуальных веществ анилин, пиридин, хинолин и их производные.
Органические амины обладают, довольно сильными основными свойствами. Поэтому в растворах минеральных кислот они практически полностью протонируются, образуя положительно заряженные ониевые соединения
RNH2 + H3 0+ ↔ RNH+3 + H2 О
Органические амины являются ПАВ катионного типа и при
адсорбции создают φ-потенциал положительного знака. Из электрокапиллярных кривых ЭКК в работах [13-16] следует также вывод о возможности некоторой адсорбции пиридиновых и
анилиновых производных и на положительно заряженной поверхности.
Степень заполнения поверхности железа этими производными не превы-
шает θ< 0,5—0,7.Для органических аминов, не имеющих дополнительных функциональных групп и сильно разветвленных радикалов, экранирование поверхности незначительно и весь эффект ингибирования можно связать с увеличением положительного значения φ-потенциала при адсорбции частиц вида RNH3+.Такой механизм действия анилиновых и пиридиновых производных доказывается также наличием корреляции между величиной Δφ определенной из ЭКК на ртути, и эффективностью торможения коррозии железа и цинка.
В присутствии поверхностно-активных анионов (Br-, I- ) адсорбция
аминов усиливается и механизм действия ингибитора нельзя уже свести только
к φ-эффекту; наряду с ним необходимо учитывать и вклад блокировки
в общее торможение.
Говоря о механизм действия производных анилина, учитывается, что эти соединения могут специфически взаимодействовать с поверхностью переходных металлов за счет π-электронов кольца и неподеленной пары электронов азота аминогруппы и давать комплексные соединения с продуктами коррозии [17, 14].
В работах Л. И. Антропова [18, 19] отмечается, что торможение коррозии ингибиторами, например, благодаря уменьшению
скорости катодного выделения водорода, может сопровождаться изме-
нением природы водородного перенапряжения. Для органических ами-
нов характерно преимущественное торможение стадии разряда, а это-
возможно лишь в случае, когда ингибитор представляет собой катио-
ноактивное ПАВ. Подобный характер влияния аминов на механизм ка-
тодного выделения водорода также подтверждает, что решающую роль
в общем эффекте торможения играют протонированные частицы вида
RNH+3, при адсорбции которых увеличивается положительное значение
φ-потенциала.
Введение различных функциональных групп в ароматические амины
и пиридины, как правило, увеличивает эффективность этих веществ в
качестве ингибиторов. Это особенно хорошо объясняется и прогнозируется на основании последовательного применения принципа линейности свободных энергий [17]. Что касается алифатических аминов, то влияние дополнительных групп, вводимых в основную цепь, невелико.Алкилэтилендиамины, алкил- и алкенилдиэтиламины и другие по-
добные амины, содержащие шесть и более атомов углерода в цепи, как
ингибиторы кислотной коррозии стали проявляют довольно высокие за-
щитные свойства, не зависящие от электронодонорных свойств аминов,
их основности и площади, экранируемой одной молекулой при адсорб-
ции [91]. Наличие кратной С=С-связи усиливает защитное действие
аминов, но не благодаря изменению основности, т. е. способности об-
разовывать аммониевые формы, а вследствие появления у таких аминов
склонности к полимеризации и поликонденсации, улучшения их адсор-
бируемости.
Протонированные формы аминов могут непосредственно участвовать в катодном процессе, ускоряя его[20].Ингибирование катодного выделения водорода за счет адсорбции амина и создания в кислых средах ионами аммония положительного φ-потенциала, а также ускорение процесса за счет каталитического разряда ионов водорода, входящих в состав RNH+3, могут либо взаимно компенсироваться, либо приводить к эффектам торможения или стимулирования.Это зависит, в частности, от микроструктуры железа и сталей [21]. Микроструктура определяет энергетическое состояние поверхности и адсорбционную способность стали по отношению к частицам RNH2 и RNH3 [22].Так, например, разряд иона аммония может быть либо стадией каталитического процесса
RNH+3 + е- →RNH2 +½H2
либо стадией образования адсорбционного слоя ингибитора
RNH+3+ е- →RNH2адс+ ½H2
Если адсорбционные силы, возникающие за счет передачи неподе-
ленной пары электронов металлу, достаточно велики, то процесс идет
по пути (5.3) и за счет блокировки поверхности комплексом RNH2адс
ускорения катодного процесса не наблюдается. Этот случай наиболее
характерен для железа и металлов его группы, атомы которых имеют
вакансии в d-зоне. На непереходных металлах адсорбция по такому
механизму менее вероятна и процесс идет по пути (5.2). Ускорение или
ингибирование определяются соотношением вклада φ- и каталитиче-
ского эффектов.
Однако реакция (5.3) не объясняет эффекта усиления ингибирова-
ния в присутствии сероводорода или ионов сульфида. Поэтому авторы
работы [22] предлагают следующий механизм ингибирования аминами
коррозии железа в кислых средах в присутствии сероводорода. Вначале
идет реакция (5.1) в объеме раствора. Далее ион аммония разрушается
с образованием адсорбированных атомов водорода и амина
RNH+3+ е- →RNH2адс + Надс
Эта реакция не является стадией каталитического процесса. Она
предполагает конкуренцию между адсорбированными молекулами ами-
на и поверхностью металла за присоединение водорода. Благодаря вы-
сокой энергии связи Fe—Haac, протону энергетически выгоднее перейти от иона аммония к поверхности металла. Нейтральная молекула амина остается в виде адсорбционного комплекса, связывается с металлом за счет неподеленной пары электронов азота и больше не подвергается протонизации. Адсорбция сульфид- или гидросульфид-ионов (FeHSадс ), через которую идет растворение металла, уменьшает энергию связи Fe—Надс и приводит к адсорбции ионов RNH+3. Благодаря этому и наблюдается неоднократно отмеченный синергизм при одновременной адсорбции поверхностно-активных катионов и анионов.
В серии работ И. Д. Вдовенко с сотрудниками [23-25] развиваются представления о важной роли образования ассоциатов молекул растворителя (воды), анионов и четвертичных аммониевых солей в проявлении последними ингибирующих свойств. Авторы обосновывают предположение о взаимодействии между ионами, входящими в состав четвертичных солей, и раствора, что приводит к изменению состояния гидратных оболочек анионов. Тетраалкиламмониевые соли упорядочивают структуру воды, образуют с ней клатратоподобные ассоциаты, дегидратируя тем самым анионы. По мере уменьшения энергии дегидратации анионов в ряду SO2-4, Сl-, Вг-, I- происходит усиление защитного действия катионов четвертичных солей аммония. Частично дегидратированные галогенид-ионы, входящие в состав ассоциатов, хемосорбируются на поверхности металла, вытесняя ранее адсорбированную воду. Блокирование таким ассоциатами поверхности препятствует протеканию коррозионного процесса. Не исключено также торможение за счет энергетического барьера, возникающего вследствие создания дополнительного положительного φ-потенциала.
Образование на поверхности прочных комплексов либо из аминов
пли ионов аммония и продуктов коррозии, либо из компонентов раствора также может быть причиной торможения коррозии.Значительно менее изучены как ингибиторы кислотной коррозии ор-
ганические нитросоединения [14], которые являются ингибиторами атмосферной коррозии и коррозии в нейтральных средах. Нитросоединения ускоряют катодный процесс при коррозии в основном за счет того, что в реакцию восстановления вступают нитрогруппы. В случае пассивирующихся металлов (например, Ti) благодаря смещению потенциала Екор в положительном направлении при наличии ингибитора это приводит к пассивации и торможению коррозии.
В случае железа и металлов его подгруппы потенциал пассивации
не достигается, а наблюдаемое торможение анодного процесса связано
с адсорбцией нитросоединений и блокировкой поверхности [101]. При
анодной поляризации металлов нитрогруппа химически не изменяется.
Полинитросоединения более эффективны, чем мононитросоединения за
счет усиления адосорбции всеми группами. Адсорбция нитросоединений носит характер специфического взаимодействия и, с учетом электроноакцепторных свойств нитрогрупп, осуществляется по механизму обратной координации. При этом электроны внешних подуровней металла переходят на орбитали адсорбата.В данных сое-
динениях адсорбционно-активным центром является наиболее отри-
цательно заряженный фрагмент молекул. Не исключено, что по меха-
низму обратной координации адсорбируются и ароматические или
ненасыщенные амины. Эти соединения могут принимать электроны на
антисвязывающие орбитали, т.е. проявлять электроноакцепторные
свойства.
1.2.2 Соединения с кратными связями
Непредельные соединения являются эффективными ингибиторами кислотной коррозии [26, 17, 14, 27].Их защитное действие значительно выше, чем у аналогов, не имеющих кратных связей. Наличие кратных связей с подвижными π-электронами обусловливает высокие адсорб-ционные и защитные свойства ацетиленовых соединений. Этиленовые соединения менее эффективны, чем ацетиленовые.
Наиболее высокими защитными свойствами обладают ацетиленовые
соединения, имеющие концевую тройную связь. Вначале это правило
было установлено для ацетиленовых углеводородов, далее для ацети-
леновых спиртов. Оно распространяется и на другие ацетиленовые
соединения (амины, эфиры, галогениды).
Уже первые исследователи ацетиленовых и этиленовых ингибиторов
обнаружили, что концентрация таких ПАВ со временем падает. Это не-
сомненно связано с химическими превращениями добавок, которые
могут протекать как на поверхности защищаемого металла, так и в
объеме раствора.
Оказалось [28], что порядок реакции химического превращения не-
предельных соединений в контакте с корродирующим металлом может
быть нулевым. Это дает основание для вывода, что такие превращения
происходят на гетерогенной поверхности металла. Надо добавить, что
нулевой порядок по веществу свидетельствует о большом заполнении
поверхности адсорбатом, что возможно только при хорошей адсорбции
ПАВ.Несмотря на высокие адсорбционные свойства ацетиленовых про-
изводных, сильная катодная пли анодная поляризация приводит к де-
сорбции как исходного вещества, так и продуктов его химического превращения[29].Одновременно необходимо отметить, что ацетиленовые соединения на переходных металлах удерживаются в широкой области потенциалов, которая значительно шире, чем при адсорбции аминов. Адсорбционные слои ацетиленовых спиртов, снижая скорость, не влияют на кинетические параметры катодного выделения водорода. Анодный же процесс, по всей вероятности, изменяет свой механизм. По-видимому, ацетиленовые производные или продукты их превращения блокируют не всю поверхность металла, а только наиболее активные центры.
Адсорбционная и ингибирующая эффективность ацетиленовых соединений зависит от заместителей, окружающих тройную связь, их полярных свойств, разветвленности. Поэтому помимо ацетиленовых углеводородов и спиртов в качестве ингибиторов используют производные, содержащие, наряду с кратными связями, другие адсорбционно-активные группы. Например, пропаргиламин более эффективен, чем пропаргиловый спирт [30]; еще более эффективен дипропаргиловый эфир. Введение заместителей большого размера в α-положение к тройной связи вызывает ее экранирование, что приводит к снижению защитных свойств. Именно по этой причине третичные спирты с большими радикалами менее эффективны, чем вторичные, а тем более первичные [31].
Высокие защитные свойства имеют также пропаргиловые эфиры
замещенных фенолов. Среди них, однако, наиболее эффективен эфир
незамещенного фенола. В этом соединении система π-связей поляри-
зуется больше по сравнению с пропаргиловым спиртом, что объясняется сопряжением π-связей бензольного кольца, непосредственно связанного с атомом кислорода. Пропаргиловый эфир бензилового спирта уже хуже ингибирует коррозию, так как эффект сопряжения здесь ослаблен. Введение в кольцо алкильных радикалов также ухудшает ингибиторные свойства эфиров. Это связано как со стерическим эффектом экранирования тройной связи, так и с понижением поляризации π,π-связей.Электроноакцепторные заместители, усиливающие поляризацию тройной связи, усиливают и ингибирующие, и адсорбционные свойства ацетиленовых эфиров [ПО]. Однако некоторые электроноакцепторные заместители (—СООН; —S03 H) не приводят к усилению ингибирования, что связано с кислотным характером этих групп.
Механизм адсорбции непредельных соединений на металлах: Металлы подгруппы железа характеризуются вакансиями в d-зоне и имеют тенден-
цию к заполнению этих вакансий электронами адсорбата. Соединения
с ненасыщенными связями в процессе адсорбции из газовой фазы обра-
зуют я-комплексы при суммарном переносе электронов от ПАВ к ме-
таллу [32]. Не исключено и образование связи по типу ковалентной.
Все сказанное выше имеет место и при адсорбции ненасыщенных
ПАВ из растворов [32]. Ацетиленовые комплексы, образующиеся при
адсорбции С=С-соединений, стабилизируются на поверхности как
π-связями, так и водородными связями [33]. Адсорбция непредельных
ингибиторов на поверхности переходных металлов может происходить
не только за счет донорно-акцепторного взаимодействия, когда элек-
троны π-связи переходят на вакантные орбитали металла. Определен-
ную роль в адсорбции должен играть и такой механизм взаимодействия
ингибиторов с поверхностью металла, когда электроны переходят на
вакантные орбитали С = С- или С == С-связей.
Анализируя особенности ингибирующего действия ацетиленовых и
этиленовых производных, можно представить себе следующий меха-
низм. Наличие π-связей придает непредельным соединениям поверхностную активность. Независимо от механизма адсорбции на переходных металлах он носит характер специфического, хемосорбционного взаимодействия. Образовавшийся адсорбционный слой ПАВ экранирует поверхность и тормозит коррозионный процесс. В зависимости от каталитических свойств металла и строения ацетиленовых ПАВ может либо наблюдаться дальнейшее химическое превращение адсорбированного ингибитора, либо не наблюдаться. В -первом случае (переходные металлы, концевая С=С-связь, сопряжение ее с другими π-связями)происходят сложные адсорбционно-полимеризационные процессы, адсорбционный слой превращается в фазовый, полимолекулярный, поверхность металла практически полностью блокируется и достигается чрезвычайно высокая степень защиты. Во втором случае (тройная связь в середине молекулы или адсорбция соединений с концевой С=С-связью на непереходных металлах) процесс ограничивается созданием адсорбционных защитных слоев.Не исключены химические превращения ацетиленовых соединений и в объеме раствора с образованием различных, как правило, плохо растворимых продуктов конденсации, полимеризации, гидрогенизации. В этом случае эффективность защитного действия добавок невысока.
Этиленовые соединения, вероятно, не подвергаются столь глубоким
химическим превращениям. Их адсорбция в отсутствие дополнительных активных групп ограничивается созданием лишь адсорбционных защитных слоев, в связи с чем эффективность таких ингибиторов ниже, чем ацетиленовых.
Процессы полимеризации и конденсации характерны также для
ПАВ, имеющих кратные связи между атомами углерода и азота [34].
1.2.3 Кислородсодержащие соединения
В качестве ингибиторов кислотной коррозии из числа кислородсодержащих соединений описаны предельные спирты и их производные, фенол и его замещенные, альдегиды и кетоны, эфиры, карбоновые кислоты [14, 17, 23, 27, 28].
Предельные спирты обладают слабой адсорбционной способностью
на железе и металлах его подгруппы [31], поэтому их ингибирующее
действие невелико.Адсорбция спиртов на железе и никеле в кислых средах обратима, хотя и носит характер специфического взаимодействия за счет полярной ОН-группы, несущей отрицательный заряд. Адсорбируемость и защитные свойства спиртов растут в ряду С3 —С6. В го же время на железе и ртути спирты С1 —С4 адсорбируются только на отрицательно заряженной поверхности в результате кулоновского взаимодействия углеводородного радикала, несущего положительный заряд, и металла [35].
Емкостные и поляризационные измерения на железе в сульфатных
растворах [36] показали значительную поверхностную и ингибирую-
щую активность спиртов С4 —С6. Рост длины углеводородной цепи и ее
разветвление ухудшают адсорбционные и ингибирующие свойства спиртов. Это можно объяснить с учетом предположения об адсорбции спиртов в виде положительно заряженной частицы и возникновения адсорбционного φ-потенциала положительного знака.
В присутствии галогенид-ионов наблюдается синергизм, который усиливается в ряду С1- > Вг- > I-.Синергизм объясняется ослаблением
гидратации галогенид-ионов в условиях стабилизации структуры воды
спиртами. Это приводит к усилению адсорбции галогенидов, улучшению экранирования поверхности и возрастанию ингибиторного эффекта.
Отмечают, что характер адсорбционного взаимодействия предельных спиртов с поверхностью, переходных металлов зависит от состояния поверхности, способа ее подготовки, предварительного контакта с тем или иным растворителем, электродного потенциала и даже времени адсорбции [31].При определенных условиях предельные
алифатические спирты могут адсорбироваться с образованием прочно
связанных с поверхностью продуктов распада. Введение в спирты до-
полнительных функциональных групп, а также переход к ароматиче-
ским оксипроизводным [16] усиливают адсорбцию н ингибиторные
свойства. Как было показано выше, ацетиленовые и этиленовые спирты
проявляют весьма высокие ингибиторные свойства.
Значительно большую ингибирующую эффективность при коррозии
железа в кислотах проявляют альдегиды. Бензальдегид и его произ-
водные обладают уже достаточно высокими ингибирующими свойствами[17, 18, 37]. Промышленное применение из ингибиторов этой группы имеет пока только формальдегид [22]. Кетоны с алифатическими радикалами уступают в эффективности альдегидам. Кетоны, один из радикалов которых является ароматическим, несколько более эффективны[25,26].
Уксусный альдегид можно считать проингибитором или ингибитором вторичного действия, так как высоким защитным свойством обладают в основном продукты его превращения. Частичное осмоление альдегида происходит и в объеме раствора, в котором находятся галогенид-ионы. В серной кислоте альдегид не подвергается превращениям, и поэтому малоэффективен как ингибитор. Отмечается, что уксусный альдегид, как ингибитор, а вернее продукты его химического превращения, проявляет синергизм с азотсодержащими ПАВ катион-
ного типа. Основания Шиффа, полученные взаимодействием различных алифатических и ароматических альдегидов и аминов, значительно активнее, чем исходные вещества, тормозят коррозию металлов [38,39]. Не исключено, что при использовании смеси аминов с альдегидами в качестве ингибиторов коррозии каталитически активных переходных металлов на их поверхности образуются основания Шиффа, чем и объясняется отмеченный выше синергизм.
Карбоновые кислоты являются слабыми ингибиторами коррозии [34].Механизм действия органических кислот в качестве ингибиторов можно, вероятно, свести к двум случаям. Так, судя по результатам работы[23], кислоты с длинной углеводородной цепью являются ингибиторами блокировочного типа. Кроме того, замедление анодного процесса в присутствии карбоновых кислот может быть связано с улучшением пассивируемости металлов. В тех случаях, когда органические кислоты способны образовывать комплексные соединения с продуктами коррозии, ингибирующие свойства зависят от прочности этих комплексов и их адсорбируемости на поверхности металла.
1.3 Растворение и пассивация железа в щелочных растворах
В работе Б.Н.Кабанова и А.И.Лейкиса [40] показывается, что процесс анодного окисления железа в щелочной среде Fe→Fe(OH)2 идет при промежуточное соединении HFeO-2. Этот процесс не может идти бесконечно долго и заканчивается вследствие пассивации железа. В настоящей работе изучался механизм процесса Fe→Fe(OH)2 , так и природа пассивации железа по отношению к этому процессу изучается зависимость перенапряжения и выхода процесса Fe→Fe(OH)2*, т.е. то количество электричества, которое расходуется на процесс до момента превращения его вследствие пассивации
активности электрода по отношению к данному процессу, оптимальной концентрации щелочи и плотности тока, а также оптимальное присутствие индифферентного иона SO2-4. В работе показывается, что выход процесса Fe→Fe(OH)2 при концентрациях от 0,05 до 3,5н возрастает линейно с увеличением концентрации щелочи, а также возрастает линейно с уменьшением плотности тока. Перенапряжение анодного электрохимического процесса ηА почти не зависит от концентрации щелочи, а также и от присутствия индифферентного иона и увеличение линейно lgi. Процесс Fe→Fe(OH)2 состоит из неравновесного перехода железа в HFeO-2 с по-следующем выпадением из раствора рыхлого пористого осадка Fe(OH)2, слабо связанного с металлом.
Различие скорости растворения в кислотах и достаточно концентрированных щелочах, а также пассивация, заставляет предположить, что в щелочах основную роль играет реакция железа с гидроксимными ионами, которая облегчена большой их концентрацией в растворе. Можно принять, что электрохимическая реакция гидроксида с железом является промежуточной стадией растворения железа в щелочах, определяющей скорость процесса. Присутствие гидроксильных ионов способствует, с одной стороны, ускорению процесса растворения, а с другой — пассивации железа. Таким образом ОН- в анодном процессе играет двойную роль. Продуктом анодного растворения являться некоторый электрохимический активный поверхностный окисел, который далее превращается в HFeO-2. На поверхности железа при определенном потенциале имеется активный адсорбционный слой, очевидно, содержащий кислород. По-видимому, это соединение и является промежуто-ным при анодном растворении железа. Некоторые подобные воззрения высказывал Эшлер[41]. Он указывает, что платина растворяется не путем прямого перехода в гидратированный ион, а посредством образования на поверхности платины комплекса с хлором и с последующим переходом комплекса в раствор. Кислород играет туже роль при растворении железа, что и Сl2 при растворении платины.
Указывается, что анодное растворение железа идет быстрее чем образование пассивирующего слоя, поэтому пассивирующий окисел должен постоянно удаляться с поверхности вместе с растворяющимся слоем железа. Показывается, что при повышении потенциала, происходит в результате увеличения плотности тока или уменьшением концентрации щелочи, выход процесса уменьшается, следовательно, с повышением потенциала процесс пассивации ускоряется больше, чем процесс растворения. Это может быть в случае, если на элементарный процесс, ведущий к пассивации, затрачивается больше электронов, чем на элементарный процесс, ведущий к растворению металла. Это можно ожидать, если валентность железа в пассивированном окисле выше двух.
Гидроксид кальция Са(ОН)2 является эффективным, безвредным и легкодоступным ингибитором коррозии, углеродистых сталей в водных средах[42,43]. В работе[44] авторами изучается возможность исполь-зования Са(ОН)2 для предупреждения питтинговой коррозии и корро-зионного растрескивания нержавеющей стали.Образцы длиной 50 мм помещали в сосуды с технической водой, используемой на объекте. В воду вводили 0,8 г/л Са(ОН)2 («х.ч.»). Для сравнения, часть образцов выдерживали в неингибированной технической воде. После извлечения образцов способность Са(ОН)г формировать на стали защитные пленки и эффективность их защитного действия оценивали визуально и методом анодного заряжения поверхности. Последнее проводили током 1 мкА/см2 в 0,1 н. растворе NaCl [43], сравнивая с образцами, выдержанными в обычной технической воде.Близость параметра пассивности к единице указывает на достаточно высокие защитные свойства пленок.Как следует из результатов, своеобразие действия Са(ОН)2 как ингибитора питтингообразования нержавеющей стали заключается в том, что, хотя при всех концентрациях Са(ОН)2 состояние стали сдвигается в пассивную область, не существует прямой зависимости между величиной удельного поляризационного сопротивления и концентрацией Са(ОН)2 в растворе.
Стабилизация защитных свойств в отношении питтинговой коррозии
нержавеющей стали отмечена в растворах, содержащих 0,8 г/л Са(ОН)2 и
более. Эти концентрации могут быть рекомендованы для защиты от пит-тинговой коррозии.
2 Экспериментальная часть
2.1 Электрохимические методы анализа
Электрохимические методы анализа – совокупность методов качественного и количественного анализа, основанных на электрохимических явлениях, происходящих в исследуемой среде или на границе раздела фаз и связанных с изменением структуры, химического состава или концентрации анализируемого вещества. Электрохимические методы анализа делятся на пять основных групп: потенциометрию, вольтамперометрию, кулонометрию, кондуктометрию и диэлектрометрию.
Потенциометрия объединяет методы, основанные на измерении эдс обратимых электрохимических цепей, когда потенциал рабочего электрода близок к равновесному значению. Потенциометрия включает редоксметрию (группа методов количественного химического анализа, основанных на применении окислительно-восстановительных реакций), ионометрию и потенциометрическое титрование.
Вольтамперометрия основана на исследовании зависимости тока поляризации от напряжения, прикладываемого к электрохимической ячейке, когда потенциал рабочего электрода значительно отличается от равновесного значения. По разнообразию методов вольтамперометрия – самая многочисленная группа из всех. Электрохимические методы анализа, широко используются для определения веществ в растворах и расплавах (например, полярография, амперометрия).
Кулонометрия объединяет методы анализа, основанные на измерении количества вещества, выделяющегося на электроде в процессе электрохимической реакции в соответствии с законами Фарадея. Различают потенциостатическую и гальваностатическую кулонометрию, причём последняя включает прямой и инверсионный методы, электроанализ и кулонометрическое титрование.
К кондуктометрии относятся методы, в которых измеряют электропроводность электролитов (водных и неводных растворов, коллоидных систем, расплавов, твёрдых веществ). Кондуктометрический анализ основан на изменении концентрации вещества или химического состава среды в межэлектродном пространстве; он не связан с потенциалом электрода, который обычно близок к равновесному значению. Кондуктометрия включает прямые методы анализа (используемые, например, в солемерах) и косвенные (например, в газовом анализе) с применением постоянного или переменного тока (низкой и высокой частоты), а также хронокондуктометрию, низкочастотное и высокочастотное титрование.
Диэлектрометрия объединяет методы анализа, основанные на измерении диэлектрической проницаемости вещества, обусловленной ориентацией в электрическом поле частиц, обладающих дипольным моментом. Методы диэлектрометрии применяют для контроля чистоты диэлектриков, например для определения малых количеств влаги [45].
Явления поляризации могут быть и вредны, и полезны. Например, при электролизе они повышают расход электроэнергии, а при работе гальванического элемента понижают отдачу электроэнергии; зато при коррозии могут вести к торможению нежелательных процессов [46].
Потенциодинамический метод получил широкое распространение, как наиболее удобный метод изучения кинетики электродных процессов. Он заключается в получении и анализе анодных и катодных поляризационных кривых, то есть установление зависимости силы поляризующего тока от заданного значения потенциала. Установив для ряда значений потенциала соответствующие значения силы тока, строят кривые, отражающие зависимость плотности тока от потенциала, т.е. потенциодинамические кривые катодной и анодной поляризации. Снятие поляризационных кривых при изучении коррозионных процессов преследует различные цели. К ним можно отнести изучение кинетики катодного и анодного процессов, установление оптимальной величины защитного тока при применении катодной защиты, графический метод расчета скорости коррозии, исследование явления пассивности и другие.
В качестве альтернативных методов коррозионных исследований применяются также потенциостатический и гальваностатический методы. Методы различаются по способу снятия поляризационных кривых. Задавая и поддерживая постоянное значение плотности внешнего тока электрода, и фиксируя, устанавливающийся при каждой плотности тока значения потенциала, получают так называемую гальваностатическую кривую.
Если наоборот, поддерживают значение потенциала и измеряют устанавливающуюся плотность тока, то получают потенциостатическую кривую.
Наиболее простым прибором для снятия потенциодинамических поляризационных кривых является потенциостат. По мнению исследователей, потенциостат обладает ценными достоинствами: низкой стоимостью, высокой точностью, простой схемой.
2.2 Методика потенциометрического определения
Перед началом проведения потенциометрических измерений был выполнен анализ состава образца антигололедного покрытия — галита разными методами определения. Была проведена математическая обработка результатов анализа, которые были табулированы в табл.1
№ п/п | Определяемый компонент | W,% | Метод определения | |
| Сухое вещество | С учетом влаги | |||
| 1 | Влажность | 8,50 | Гравиметрический | |
| 2 | Нерастворимый остаток | 2,80 | 2,58 | Гравиметрический |
| 3 | NaCl | 92,7 | 85,44 | Пламенно-фотометрический |
| 4 | K2 SO4 | 0,45 | 0,41 | Пламенно-фотометрический |
| 5 | MgSO4 | 2,30 | 2,13 | Титриметрический, турбидиметрический |
| 6 | CaSO4 | 0,75 | 0,69 | Титриметрический, турбидиметрический |
| 7 | Карбонаты | Примеси<1% | <<1% | Титриметрический |
| 8 | Нитраты | — | — | Фотометрический |
| 9 | Соли Fe(III) | Примеси<<1% | <<1% | Фотометрический с салициловой кислотой |
(таблица1)
Проведение потенциометричких измерений
Метод основывается на пассивации стали Ст85 в агрессивной среде (галите), и заключается в оценке защитного действия растворов солей ингибиторов и их смесей по отношению к образцу стальной арматуры путем сравнения данных, полученных при измерении изменения плотности электрического тока в зависимости от изменения потенциала (потенциодинамический метод) или наоборот – изменения потенциала в зависимости от изменения плотности электрического тока, пропускаемого через образец (гальванодинамический метод).
Электрохимический метод испытания можно применять для:
1. определения наиболее оптимальных растворов солей ингибиторов для защиты арматурной стали Ст85 от действия антигололедного реагента(галита).
2. оценки и сравнения влияния различных концетраций растворов солей в тройных системах «галит-мочевина-гидроксид кальция» и «галит-аммиачная селитра-гидроксид кальция» по отношению к стальной арматуре.
В зависимости от приборов, применяемых для измерения параметров электрического тока, испытания можно проводить одним из двух способов: потенциодинамическим и гальванодинамическим.
Анодные кривые снимались в соответствии с методикой [47] в потенциодинамическом режиме на потенциостате П–50-1 с регистрацией сигналов «потенциал-ток». Электрохимические измерения проводились в стандартной стеклянной трёх электродной ячейке с нераздельным катодным и анодным пространством. Анодом служит исследуемый металл, катодом – платиновый электрод (вспомогательный электрод), электродом сравнения – хлорсеребряный электрод. Электродные потенциалы, измеренные по отношению к хлорсеребряному электроду, можно пересчитать на нормальную водородную шкалу по формуле:
φн.в.э. = φх.с.э. + φ
где φ – измеренный потенциал,
φх.с.э. – потенциал хлорсеребряного электрода (φх.с.э. = + 0,222 В при t= 20°С)
Эксперимент проводился в свежем растворе электролита при комнатной температуре. Подготовленный образец стали Ст85 устанавливали в электрохимическую ячейку и определяли величину стационарного потенциала. При снятии поляризационной кривой потенциодинамическим методом с помощью потенциостата в автоматическом режиме поляризовали образец со скоростью 13 В/ч, измеряя величину тока через каждые 0,03 В изменения потенциала.
Рабочая поверхность исследуемого электрода измерялась в сантиметрах квадратных с точностью до 0,01 см2 .
Результаты эксперимента представлены в виде графических зависимостей: φ = f (lgi )
где φ – потенциал, В;
i – плотность тока А/см2
2.3 Подготовка электродов для электрохимических измерений
Электродами называют части проводников гальванической цепи, погруженные в вещества (электролиты), подвергаемые действию электрического тока. Электроды делают чаще всего из твердых, проводящих ток веществ, т. е. из металла или угля. Жидкие электроды встречаются нередко в лабораторной и заводской практике, примером чему могут служить ртутные электроды, а также электроды из других расплавленных металлов. Термин электрод предложен Фарадеем, чтобы им заменить для частных случаев более общий термин «полюсы». Отсюда следует, что характер электрода может быть положительного полюса; такой электрод Фарадей назвал анодом, а электрод характера отрицательного полюса получил название катода. В зависимости от тех химических превращений, которые совершаются при прохождении тока на границе электрод/электролит, электроды бывают обратимые и необратимые. Границу эту принято графически обозначать вертикальной чертой, как и вообще границу двух веществ, на которой могут развиваться электровозбудительные силы.
Обратимым электродом называют такой, у которого в месте соприкосновения электрода с электролитом при перемене направления тока совершается химическое прекращение, как раз обратное тому, что совершалось при первоначальном направлении тока. Электроды, не удовлетворяющие этому требованию, носят название необратимых. Пример обратимого электрода: тяжелый металл (медь цинк, кадмий и др.), погруженный в раствор соли того же металла. Кроме качественных требований, обратимый электрод часто должен удовлетворять количественным требованиям. Такой случай наблюдается для газо-платиновых электродов, т. е. для платины, погруженной частью в раствор электролита, частью же в атмосферу газа, выделяющегося при электролизе, хотя бы, например, в атмосферу водорода. Если сила обратного тока будет такова, что у водород – платинового анода будет происходить только растворение водорода, но не будет выделения кислорода, такой электрод обратим для водород – платинового катода. Обратимые металлические или газо – металлические электроды носят название электродов первого рода. Электроды первого рода обратимы для катионов Сu2+, Zn2+, Cd2+, H+ и т. д., а газо – металлические — для, Сl- и др. Электроды второго рода являются обратимыми для анионов Сl-, Вr-, I- и др. На существование обратимости в этих электродах было впервые указано Нернстом, он же дал и теорию этих электродов. Они представляют металлы, покрытые слоем нерастворимых солей этих металлов, погруженные в раствор соли с тем же анионом, как и у нерастворимой соли. Примером может служить ртутный электрод, покрытый слоем каломели (Hg2 Cl2 ), или серебряный электрод, покрытый слоем хлористого серебра (AgCl), погруженные в раствор хлористого калия. При прохождении тока в одном направлении, когда электрод является анодом, выделяющийся ион хлора, соединяясь с металлом электрода, образует нерастворимую соль, т. е. как бы хлор «осаждается током на электроде»; когда же электрод становится катодом, хлор нерастворимой соли переходит в раствор. Эта качественная сторона явлений не дает, конечно, полной картины происходящих процессов и говорит о том, что в таком электроде хлор является как бы металлом, отличающимся только знаком электричества его иона, что важно только для общей характеристики явления. Теория же явления, дающая точное представление, основана на химическом взаимодействии веществ у электрода. Еще сложнее теория обратимых электродов 3-го рода. Эти электроды предложены Лютером, как обратимые для металлов, выделяющих водород из воды. Электроды 3-го рода состоят из металла, контактирующего с двумя труднорастворимыми солями. В результате электрохимической реакции на электроде менее растворимая соль превращается в более растворимую, а потенциал электрода определяется термодинамической активностью катионов более растворимой соли.
Форма и величина электродов бывает самая разнообразная, в зависимости от тех требований, которым они должны удовлетворять. Существенной для электрода является та его поверхность, через которую ток попадает в электролит [48].
Исходное состояние поверхности электрода во многих случаях сказывается на его коррозионно – электрохимическом поведении не менее сильно, чем составы металлов, растворов и других основных условий эксперимента, поэтому выбор способа предварительной подготовки поверхности исследуемого электрода является одним из решающих факторов при электрохимических измерениях. Применяя исходную обработку поверхности стремятся прежде всего исключить влияния царапин, вмятин, остатков окалины и других загрязнений, неравномерной шероховатости, различия качества поверхности исследуемых материалов и т.п., что приближает видимую поверхность к истинной. Выбранный способ подготовки должен обеспечить достаточную воспроизводимость опытных данных. Перед началом эксперимента рабочая поверхность электрода отшлифовывалась наждачной бумагой различной зернистости, полировалась, обезжиривалась этиловым спиртом, а затем промывалась дистиллированной водой. Перед исследованием измеряли площадь поверхности электрода. Электроды готовились из арматурной стали Ст.85 класса ВР-II.
Рабочую поверхность стального стержня S рассчитывают после снятия поляризационной кривой по формуле
S=2πrl +πr2
где r – радиус стального стержня, cм;
l – рабочая длина стального стержня, cм.
2.4 Приготовление рабочих растворов
В настоящей работе использовались следующие растворы: 20% раствор NaCl, 20% раствор (NH4 )2 CO, 20% раствор NH4 NO3, 20% раствор Ca(H2 PO4 )2 и раствор Ca(OH)2 =0,8 г/л.
Приготовление 20% растворов:
20% раствор NaCl, 20% раствор (NH4 )2 CO, 20% раствор NH4 NO3, 20% раствор Ca(H2 PO4 )2 получают растворением точно известного количества химически чистых веществ в определенном объеме воды. Рассчитанную навеску каждой соли m=200 г. предварительно взвешивают на технических, а затем порциями на аналитических весах и растворяют в мерной колбе объемом на 1000 мл. Объем раствора доводят до метки дистиллированной водой и тщательно перемешивают.
В настоящей работе использовались следующие смеси растворов солей в объемных соотношениях:
W(NH4)2CO или NH4NO3 ,% | WNaCl ,% | WCa(OH)2 ,% |
| — | — | 100 |
| 80 | 10 | 10 |
| 60 | 20 | 20 |
| 50 | — | 50 |
| 40 | 30 | 30 |
| 20 | 40 | 40 |
| — | 50 | 50 |
Объем электрохимической ячейки V=175мл (100%).
3 Экспериментальная часть и её обсуждение
3.1 Анодное поведение углеродистой стали в исходных растворах
Известно, Сl — ионы, в основном, являются активаторами коррозионного процесса[ 6-9 ].Ряд авторов придают ионам Сl — и пассивирующие свойства в больших концентрациях [ 7 ]. На рисунке 1 представлена активная поляризационная кривая ( АПК) углеродистой стали Ст.85 в 20% растворе галита ( NaCl с примесями, таблица 1). Сталь находится в области φ активного растворения. Оптимальное значение φ свободной коррозии равно 0,5 В -0,56 В где металл растворяется безпрепятственно. Наблюдается участок прямой Тафеля с незначительным углом наклона, что является свидетельством выше сказанного утверждения (рисунок 1 кривая 1). АПК стали в мочевине ( рисунок 1кривая 2) имеет электрохимические характеристики, указывающие на сдвиг φ в более положительную область с уменьшением токов растворения. Ряд авторов [ 41-44] делают ссылку на ингибирующее действие Ca(OH)2 в нейтральных средах[42]. Однако как показывает АПК стали Ст.85 токи растворения достаточно велики, хотя растворение протекает с φ в области неявно выраженной анодной активации (рисунок1 кривая 3).АПК стали в NH4 NO3 имеет токи значительнее, чем сталь в выше указанных растворах (рисунок 1 кривая 4). Испытание стали в растворах фосфатов не привели к ожидаемым результатом (рисунок1 кривая4).
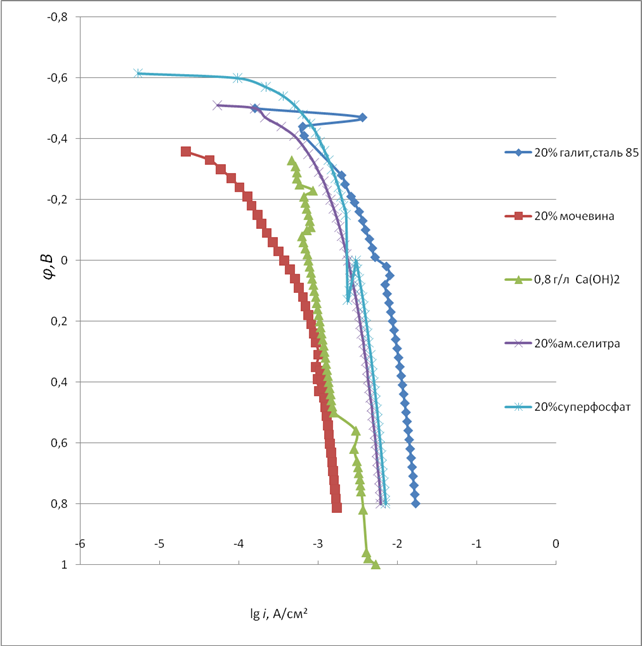
Рисунок 1– АПК стали 85 в чистых растворах.
3.2 Анодное поведение стали Ст 85 в смесях солей
Учитывая, что гидроксид кальция подщелачивает растворы с образованием на металлической поверхности защитных пленок, представлял интерес исследовать его ингибиторные свойства в антигололедных реагентах.
На рисунке 2 представлены анодные поляризационные кривые (АПК) углеродистой стали 85 в антигололедных реагентах систем «галит – мочевина — гидроксид кальция» и «галит — нитрат аммония -гидроксид кальция». На рисунке 2 потенциал свободной коррозии стали (φсв.к. ) находиться в области потенциалов активного растворения стали, но с менее отрицательным значением такового по сравнению со смесями на рисунке 3. Область активного растворения незначительна. АПК круто опускается к положительным значениям потенциалов. Скорость анодного растворения невелика со сдвигом φ в положительном направлении. Добавка в смесь 80% мочевины- 10% галита-10% гидроксида кальция сдвигает φсв.к стали в более отрицательную область потенциалов. Пики растворения возрастают (рисунок 2 кривая 2). На АПК оптимальный φсв.к регистрирует Тафелевский участок потенциалов.
С увеличением концентрации Ca(OH)2 до 20% в смеси 20% галит-60% мочевины электрохимические характеристики стали улучшаются (рисунок 2 кривая 3). Более того, неактивно возрастают токи анодного растворения оптимального φсв.к до φ≈ 0,06 В. Дальнейший сдвиг φ в положительную область потенциалов заметно сказывается на интенсивный рост токов анодного растворения.
Мочевина вносит свой вклад в ингибиторное действие гидроксида кальция очевидно ( рисунок 2 кривая 4). Потенциал свободной коррозии значительно сдвинут в область φ меньших значений по сравнению ( рисунок 2, кривая 2-6) с φсв.к в других смесях. Имеется область потенциалов пассивного состояния. В смеси 30% галит-40% мочевины-30% гидроксида кальция сталь также находится в активном состоянии во всех областях φ анодной поляризации (рисунок 2, кривая 5). Существенных различий в анодном поведении стали в сравнении с АПК в других смесях не наблюдается. Без мочевины ( 50% галит-50% гидроксид кальция) электрохимические характеристики стали ухудшаются ( рисунок 2, кривая 6).

Рисунок 2- АПК стали 85 в системе «мочевина – галит — гидроксид кальция».
Определенный интерес представляет практическое рассмотрение другой смеси солей NH4 NO3 — NaCl -Ca(OH)2 в качестве антигололедногореагента. Как показывает рисунок 3 применение NH4 NO3 вместо мочевины не улучшает электрохимические характеристики металла. Наблюдается активное растворение металла во всем интервале потенциалов анодной поляризации. Система мало эффективна.
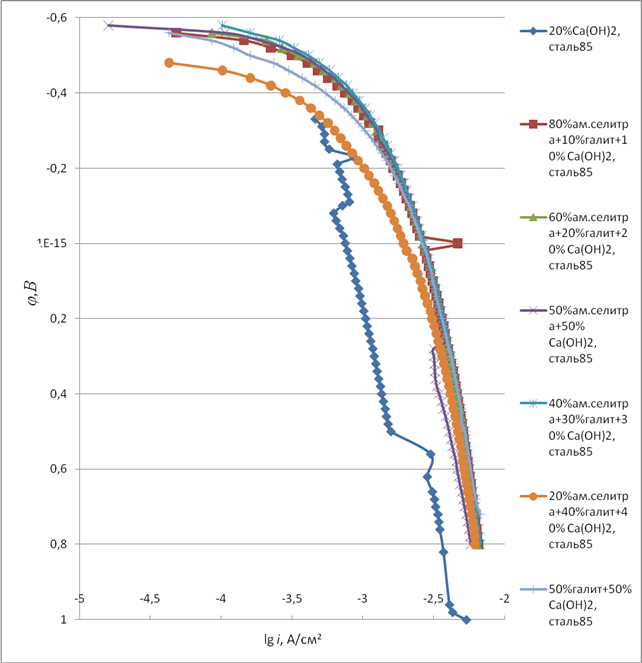
Рисунок 3- АПК стали 85 в системе «ам.селитра – галит — гидроксид кальция».
3.3 Скорость коррозии в чистых растворах
Гравиметрический метод изучения коррозии и определения скорости коррозии является наиболее широко распространенным методом количественной оценки коррозии металлов. Его достоинства – сравнительная простота реализации и получения наиболее достоверной и надежной информации о потерях в массе металла в результате коррозии.
При испытании образцов коррозионная стойкость оценивалась по внешнему виду (визуальный метод) и по массовым потерям в результате коррозии (гравиметрический метод).
Δm, (г) рассчитывали по уравнению:
![]()
где ia – плотность тока, А/см2 ;
S – площадь образца, м2 ;
АМе – атомный вес металла, г;
τ – время испытаний, час;
n – число электронов;
F – число Фарадея.
Показатель коррозии К, г/(м2. час) рассчитывали по уравнению:
![]() ,
,
Недостаток гравиметрического метода определения скорости коррозии по изменению потерь массы прокорродировавшего металла состоит в невозможности получения информации о протекании локальных форм коррозии, поскольку коррозионные потери относятся ко всей площади поверхности образца, без учета локализации коррозии.
Таблица 2
Гравиметрические показатели
| Чистые растворы | ∆m, г | K, г/м2 ·ч |
| (NH4 )CO | 0,0034 | 0,0009 |
| Ca(OH)2 | 0,0056 | 0,00017 |
| NH4 NO3 | 0,024 | 0,0045 |
| Ca(HPO4 )2 | 0,033 | 0,006 |
| NaCl | 0,596 | 0,017 |
Выводы
1. Проведены исследования электрохимического и коррозионного поведения углеродистой стали марки Ст. 85 в растворе галита (натрий хлор).
2. Исследовано коррозионное поведение углеродистой стали марки Ст. 85 в чистых растворах солей и тройных системах «мочевина – галит — гидроксид кальция», «аммиачная селитра – галит — гидроксид кальция».
3. Рассчитаны скорости коррозии (К) в исследуемых средах. Результаты измерений показали, что наименьшая скорость коррозии отмечалась в растворе мочевины.
4. На основе проведенных исследований установлено, что сталь Ст.85 имеет наилучшие электрохимические характеристики в системе «мочевина – галит — гидроксид кальция» при соотношении:50%мочевины+50%Ca(OH)2 и 40%мочевины+30%галит+30% Ca(OH)2 .
5. Опыты показали, что замена мочевины в тройной системе на аммиачную селитру не дает положительных результатов.
6. После обобщения всех данных было принято решение, что состав-ление тройных систем для защиты от коррозии в присутствии антигололедного покрытия мало эффективно и может использоваться только в комплексе с другими методами борьбе с коррозией углеродистой стали Ст.85
Список использованных источников
1. Решетников С. М. Ингибирование кислотной коррозии металлов. Ижевск: Удмуртия, 1980, 128 с.
2.Решетников С. М. — ЖПХ, 1979, т. 52, № 6, с. 1322—1325.
3. Решетников С. М. — ЖПХ, 1981, т. 54, № 3, с. 586—589.
4. Решетников С. М. — ЖПХ, 1980,
т. 53, № 3, с. 572—577.
5. ВдовенкоИ. Д., Вакуленко Л. И. — Укр. хим. журнал, 1976,
т 42, № 4, с. 436—438.
6. Томашов Н.Д. Теория коррозии и защиты металлов / Н.Д. Томашов. — М.: Изд-во АНСССР, 1959. — 592 с.2.
7. Л. Ванюкова, Б.Н. Кабанов// Докл. АН СССР. О стали 12Х18Н10Т в оборотной воде, содержащей ионы хлора,1940. Т. 14. N 12. С. 1620-1625
8. АкимовГ.В. Теория и методы исследования коррозии. М.-Л.: изд-во АН СССР, 1945, 414 с.
9. Хедже.Ингибиторы коррозии металлов. М.: Госхимиздат, 1954. 185 с
10. Annand R. R., Hurd R. М., Hakerman N. J. Electrochem. Soc 1965. v. 112.
№ 2, p. 138; 1965, v. 112, № 2, p. 144.
11. Riggs O. L., Every R. L. «Corrosion»,1962, v. 18, № 7. p. 262t.
12. Trabanelly C, Carassiti V. «Advances in Corrosion Science аnd Technology.*.Plenum Press, 1970, v. 1.
13. Антропов Л. Я., Погребова И. С. — В кн.: Коррозия и защита от коррозии. Итоги науки и техники. М.: ВИНИТИ, 1973, т. 2, с. 27—112
14. Розенфельд И. Л. Ингибиторы коррозии. М.: Химия, 1977. 352 с.
15. Дамаскин Б. Б., Петрий О. А., Батраков В. В. Адсорбция органических соединений на электродах. М.: Наука, 1968. 333 с.
16. Антропов Л. Я., Макушин Е. М., Панасенко В. Ф.
Ингибиторы коррозии металлов. Киев:. Техника, 1981. 181 с.
17. Григорьев В. Г., Экилик В. В. Химическая структура и защитное действие ингибиторов коррозии. Ростов н/Д: Ростовский университет, 1978. 164 с.
18. Антропов Л. И. Теоретическая электрохимия. М.: Высшая школа, 1984, 519 с.
19. Петренко А. Г., Антропов Л. И. — ЖПХ, 1958, т. 31, № 5, с. 949—951.
20. Подобаев Н. Я., Савиткин И. И. — Электрохимия,
1975, т. 11, № 5, с. 842—844.
21. Розенфельд И. Л., Кузнецов 10. Я., Белов А. В. — Защита металлов, 1977, т. 13, № 4, с. 448—450.
22. Розенфельд И. Л., Персианцева В. Я., Дамаскина Т. А. — Защита металлов, 1973, т. 9,№ 6, с. 687—690.
23. Вдовенко И. Д., Лисогор А. И., Пименова К. И. и др. — Укр. хим. жури., 1978, т. 44, № 6, с. 599—605.
24. Вдовенко И. Д., Перехрест Н. А., Лисогор А. И. — В кн.: Электродные процессы при электроосаждении и растворении металлов.—Киев: Наукова Думка, 1978, с. 124—129.
25. Вдовенко И. Д., Лисогор А. Я., Перехрест Н. А. — Укр. хим. журн., 1981, т. 47, № 7, с. 683—686.
26. Путилова И. #., Балезин С. А., Баранник В. П. Ингибиторы коррозии металлов. М.: Госхимиздат, 1954. 185 с.
27. Алцыбеева А. И., Левин С. 3. Ингибиторы коррозии металлов. Справочник. Л.: Химия, 1968. 264 с.
28. PutilowaJ. N., KoczanowaG. N.,LoluaA. M. e. a. — Ochr. przed. koroz., 1970, № 8, s. 1—3.
29. Подобаев H. Я., Воскресенский А. Г. — ЖПХ, 1970, т. 43, c. 834—838.
30. Гидаянес К., Иванов Е. С, Караев С. Ф., Мовсум-заде М. М. — Изв. вузов. Нефтьигаз,.1982, № 3, с. 52, 88.
31. Caprani A., Morel Я/г.— In: 5th Eur. Symp. Corros. Inhibit., Ferrara, 1980, v. 2, p. 471—478.
32. Закумбаева Г. Д. Взаимодействие органических соединений с поверхностью металлов VIII группы. Алма-Ата: Наука, 1978. 304 с.
33. TedeschiR. G. — Corrosion (USA), 1975, v. 31, № 4, p. 130—134.
34. Путилова И. Я., Числова Е. Я., Лолуа А. М. — В кн.: Ингибиторы коррозии металлов. М.: МГПИ им.В. И. Ленина, 1969, с. 40—50.
35. BanerjecS. N… Guha B. R. — J. Indian
Chem. Soc, 1979, v. 56, № 9, p. 880—884.
36. Кичигин В. #., Шадрин О. А., Шерстобитова И. Н. Влияние адсорбированных алифатических спиртов на кинетику разряда
ионов водорода на железном электроде. Рук. деп. вВИНИТИ, М., № 1057—82 Деп.12 с.
37. Foroulis Z, А. — In: 4th Eur. Symp. Corros.Inhibit. Ferrara, 1975, v. 3, p. 542—559.
38. Desai М. N.,Shah G. V., Pandya М. М. — In: 5th Eur. Symp. Corros. Inhibit., Ferrara, 1980, v. 2, p. 397—403.
39. Подобаев H. И., Харьковская H. Л. — В кн.: Ингибиторы коррозии металлов. М.: МГПИ им. В. И. Ленина, 1972, с. 110—116.
40. Кабанов Б.Н., Лейкис А.И., ЖФХ, 20,9, 1946 г.
41. Эшлер Б.В., ЖФХ,18,131,1944 г.
42. Харитонов Ю. Я., Аколъзин А. П., МорозоваЛ. Н. А.с. СССР№ 939594.- Опубл. в Б.И. 1982, Л» 24.
43. Аколъзин А. П., Пхулчанд Гхош, Харитонов Ю. Я. Защита металлов, 1983, т. 19, № 2, с. 302.
44. Розенфельд И. Л., Данилов И. С. В кн.: Новые методыисследования коррозии металлов. М.: Наука, 1973, с. 193.
45. Новаловский, В.М. К развитию электрохимической теории коррозионых процессов в программе научно–технического сотрудничества стран – членов СЭВ / В.М.Новаковский // Защита металлов. – 1979. – №1. – С. 3–16.
46. Лобанов, И.А. Долговечность армоцемента / Доклады ХХIII научной конференции ЛИСИ, 1965 г
47. Розенталь, Н.К. Коррозионностойкие бетоны особо малой проницаемости / Н.К.Розенталь, Г.В.Чекий. // Бетон и железобетон. – 1998. – «2. –С. 27 – 29.
48. Куприн, В.П. О прогнозировании адсорбции органических веществ на металлах / В.П.Куприн, Е.А.Нечаев // Защита металлов. – 1991. – №5. – С. 782 – 788.