Реферат: Химия, элементы таблицы Менделеева
Седьмая группа периодической системы
Из членов данной группы водород был рассмотрен ранее. Непосредственно следующие за ним элементы — F, Сl, Br и I — носят общее название г а л о г е н о в. К ним же следует отнести и элемент № 85 — астат (Аt). Другую часть группы составляют элементы п о д г р у п п ы м а р г а н ц а (Мп, Тс, Rе).
Как видно из приводимых электронных структур, атомы галогенов имеют 7 электронов во внешнем слое. Основываясь на этом, можно наметить некоторые черты их химической характеристики: так как до устойчивой конфигурации внешнего слоя не хватает лишь по одному электрону, наиболее типичным для галогенов должны быть соединения, в которых эти элементы играют роль о д н о в а л е н т н ы х м е т а л л о и д о в. С другой стороны, их максимальную положительную валентность можно ожидать равной семи.
Иначе обстоит дело в подгруппе марганца. Здесь незаконченными являются уже д в а внешних слоя. Так как в наиболее удаленном от ядра слое находится только два электрона, тенденции к дальнейшему присоединению электронов не будет. Наоборот, при их отдаче и образовании валентных связей могут принять участие и 5 электронов следующего слоя. Поэтому максимальную положительную валентность элементов подгруппы марганца также можно ожидать равной семи. Таким образом, по своим основным тенденциям элементы обеих групп сильно отличаются друг от друга: тогда как галоиды должны в первую очередь характеризоваться резко выраженной металлоидностью, марганец и его аналоги будут вести себя как металлы.
Фтор.
На земной поверхности фтор встречается исключительно в составе солей. Общее его содержание в земной коре равняется 0,02 %. Основная масса фтора распылена по различным горным породам. Из отдельных форм его природных скоплений наиболее важен минерал ф л ю о р и т — СаF2.
Фтор является «чистым элементом» — состоит только из атомов 19F. Впервые он был обнаружен в плавиковой кислоте (1810 г.). Попытки выделить этот элемент долгое время оставались безуспешными, и свободный фтор удалось получить лишь в 1886 г.
Основная масса фтора земной поверхности обязана своим происхождением горячим недрам Земли (откуда этот элемент выделяется вместе с парами воды в виде НF). Среднее содержание фтора в почвах составляет 0,02 %, в водах рек — 2·10-5 % и в океане — 1·10-4 %. Человеческий организм содержит фтористые соединения главным образом в зубах и костях. В вещество зубов входит около 0,01 % фтора, причем большая часть этого количества падает на эмаль [состав которой близок к формуле Са5F(РO4)3]. В отдельных костях содержание фтора сильно колеблется. Для растительных организмов накопление фтора не характерно. Из культурных растений относительно богаты им лук и чечевица. Обычное поступление фтора в организм с пищей составляет около 1 мг за сутки.
Установлено, что содержание фтора в питьевой воде сильно влияет на состояние зубов людей (и животных). Наилучшим является наличие около 1 мг фтора в литре. Содержание ниже 0,5 мг/л способствует развитию кариеса, а выше 1,2 мг/л — крапчатости эмали. В обоих случаях зубы подвергаются более или менее быстрому разрушению.
Элементарный фтор получают путем электролиза фтористых соединений. причем он выделяется на аноде по схеме:
2 F-® 2 е-+ 2 F ® 2 е + F2
Электролитом обычно служит смесь состава КF·2НF (часто с добавкой LiF). Процесс проводят при температурах около 100 'С в стальных электролизерах со стальными катодами и угольными анодами.
Удобная лабораторная установка для получения фтора показана на рис.
&&&&&&&&&& рис
Электролизу подвергают легкоплавкую смесь состава КF·3НF, помещенную в служащую катодом внешний медный сосуд А. Анод из толстой никелевой проволоки помещается в медном цилиндре Б, нижняя боковая часть которого имеет отверстия. Выделяющийся фтор отводится по трубке В (а водород — через отвод Г). Все места соединения отдельных частей прибора делают на пробках из СаF2 и замазке из РbО и глицерина.
Свободный фтор состоит из двухатомных молекул и представляет собой почти бесцветный (в толстых слоях зеленовато -желтый) газ с резким запахом. Он сгущается в светло-желтую жидкость при -188 °С и затвердевает при -220 °С.
Критическая температура фтора равна -129 °С, критическое давление 55 атм. При температуре кипения жидкий фтор имеет плотность 1,5 г/см3, а теплота его испарения составляет 1,6 кДж/моль. Жидкий фтор, как и его смесь с жидким кислородом (“флокс”), может служить энергичным окислителем реактивных топлив. С SiО2 или стеклом фтор не реагирует. При охлаждении ниже -252 °С его желтоватые кристаллы обесцвечиваются.
С химической стороны фтор может быть охарактеризован как о д н о в а л е н т н ы й м е т а л л о и д и притом с а м ы й активный из всех металлоидов. Обусловлено это рядом причин, в том числе легкостью распада молекулы F2 на отдельные атомы — необходимая для этого энергия составляет лишь 159 кДж/моль (против 493 кДж/моль для О2 и 242 кДж/моль для С12). Атомы фтора обладают значительным сродством к электрону и сравнительно малыми размерами. Поэтому их валентные связи с атомами других элементов оказываются прочнее аналогичных связей прочих металлоидов (например, энергия связи Н-F составляет — 564 кДж/моль против 460 кДж/моль для связи Н-О и 431 кДж/моль для связи Н-С1).
Связь F-F характеризуется ядерным расстоянием 1,42 А. Для термической диссоциации фтора расчетным путем были получены следующие данные:
Температура, °С | 300 | 500 | 700 | 900 | 1100 | 1300 | 1500 | 1700 |
| Степень диссоциации, % | 5·10-3 | 0,3 | 4,2 | 22 | 60 | 88 | 97 | 99 |
Атом фтора имеет в основном состоянии структуру внешнего электронного слоя 2s22p5 и одновалентен. Связанное с переводом одного 2р-элсктрона на уровень 3s возбуждение трехвалентного состояния требует затраты 1225 кДж/моль и практически не реализуется.
Сродство нейтрального атома фтора к электрону оценивается в 339 кДж/моль. Ион F- характеризуется эффективным радиусом 1,33 А и энергией гидратации 485 кДж/моль. Для ковалентного радиуса фтора обычно принимается значение 71 пм (т. е. половина межъядерного расстояния в молекуле F2).
Подавляющее большинство м е т а л л о в соединяется с фтором уже при обычных условиях. Однако взаимодействие часто ограничивается образованием плотной поверхностной пленки фтористого соединения, которая предохраняет металл от дальнейшего разъедания. Так ведут себя, например, Сr, Ni и Мg, которые поэтому оказываются практически устойчивыми по отношению к фтору (в отсутствие воды).
Так как фтористые производные м е т а л л о и д н ы х элементов обычно легколетучи образование их не предохраняет поверхность металлоида от дальнейшего действия фтора. Поэтому взаимодействие часто протекает значительно энергичнее, чем со многими металлами. Например, кремний, фосфор и сера воспламеняются в газообразном фторе. Аналогично ведет себя аморфный углерод (древесный уголь), тогда как графит реагирует лишь при температуре красного каления. С азотом и кислородом фтор непосредственно не соединяется.
От водородных соединений других элементов фтор отнимает водород. Большинство оксидов разлагается им с вытеснением кислорода. В частности, вода взаимодействует по схеме
F2 + Н2О ® 2 НF + O
причем вытесняемые атомы кислорода соединяются не только друг с другом, но частично также с молекулами воды и фтора. Поэтому, помимо газообразного кислорода, при этой реакции всегда образуются пероксид водорода и оксид фтора (F2О). Последняя представляет собой бледно-желтый газ, похожий по запаху на озон.
Окись фтора (иначе — фтористый кислород — ОF2) может быть получена пропусканием фтора в 0,5 н. раствор NаОН. Реакция идет по уравнению:
2 F2 + 2 NаОН = 2 NаF + Н2О + F2О
Молекула F2О имеет структуру равнобедренного треугольника [d(FО) = 141 пм РFОF = 103°] и небольшой дипольный момент (m = 0,30), Для средней энергии связи O-Г дается значение 192 кДж/моль.
При охлаждении до -145 °С окcид фтора сгущается в желтую жидкость (плотность 1,5 г/см3), затвердевающую при -224 °С. Критическая температура F2О равна -58 °С, критическое давление 49 атм. Жидкая оксид фтора смешивается в любых соотношениях с жидкими O2, F2, O3 и способен растворять большие количества воздуха. Несмотря на эндотермичность F2О (теплота образования 25 кДж/моль), он все же сравнительно устойчив, например еще не разлагается при нагревании до 200 °С (энергия активации термического разложения равна 171 кДж/моль). Почти неразлагается оксид фтора и холодной водой, в которой он малорастворим (7: 100 по объему при 0 °С). Напротив, в щелочной среде (или под действием восстановителей) разложение F2О идет довольно быстро. Смесь его с водяным паром при нагревании взрывается реакция идет по уравнению:
ОF2 + Н2О = 2 НF + O2 + 326 кДж
Оксид фтора является сильным окислителем и очень ядовит.
Формально отвечающая F2О, как ангидриду, ф т о р н о в а т и с т а я к и с л о т а (НОF) частично образуется при взаимодействии медленного тока фтора под уменьшенным давлением с охлаждаемой водой. Выделенная лишь в очень малых количествах (порядка мг), она представляет собой бесцветное вещество (т. пл. — 117 °С) с высоким давлением пара (5 мм рт. ст. уже при — 64 °С), в обычных условиях довольно быстро разлагающееся на НF и O2. Молекула НОF характеризуется следующими параметрами: d(ОН) = 0,96, d(ОF) = 1,44 А, РНОF = 97°. Фторноватистая кислота является, по-видимому, сильной, но водой она быстро гидролизуется, в основном по уравнению:
НОF + НОН = НF + Н2О2
Соли ее не получены, но известны вещества, которые можно рассматривать как продукты замещения ее водорода на радикалы металлоидного характера, т. е. как г и п о ф т о р и т ы этих радикалов.
Подобно самому фтору, его окись является одним из возможных эффективных окислителей реактивных топлив. Например, при использовании углеводородов СnН2n в качестве горючего и жидких F2 и F2О в качестве окислителя относительные (O2 = 1) расчетные значения удельного импульса и скорости ракет составляют 1,05 и 1,2 для F2, или 1,16 и 1,4 для F2О.
При действии тихого электрического разряда на охлаждаемую жидким воздухом смесь газообразных фтора и кислорода образуются оранжево-красные кристаллы состава F2O2 (т. пл. -163 °С). Оксид этот (дифтордиоксид) устойчив лишь ниже -80 °С, а при дальнейшем нагревании начинает распадаться на элементы. Изучавшееся при очень низких температурах взаимодействие его с различными другими веществами протекает, как правило, чрезвычайно бурно (нередко — со взрывом).
Молекула F2О2 полярна (m = 1,44) и по строению подобна молекуле перекиси водорода. Она характеризуется параметрами d(FО) = 158 пм, d(OO) = 122 пм, РООГ = 110° при угле около 88° между связями F-O. Так как d(OO) в молекуле O2 равно 121 пм, можно думать, что присоединение к ней двух атомов фтора существенно не искажает ее внутреннюю структуру (т. е. что дифтордиоксиду отвечает формула F-O-O-F с четырехвалентными атомами кислорода). Энергии связей OO и ОF оцениваются соответственно в 564 и 75 кДж/моль.
Гомологами F2О и F2О2 являются окислы фтора общей формулы F2Оn, где n = 3, 4, 5, 6. Они были получены действием тихого электрического разряда на смеси фтора с кислородом при температурах порядка -200 °С и под сильно уменьшенным давлением (например, синтез F2О6 велся при -210 °С и давлении около 1 мм рт. ст.). Все эти полипероксиды фтора представляют собой жидкие или твердые коричнево-красные вещества, устойчивые лишь при очень низких температурах (например, F2О6. — ниже -200 °С) и являющиеся чрезвычайно сильными окислителями. Интересно, что F2O3 нерастворим в жидком O2 или F2 (отличие от F2O2).
Практическое использование свободного фтора развилось сравнительно недавно. Потребляется он главным образом для фторирования органических соединений (т. е. замены в них водорода на фтор).
Процесс этот приобрел большое значение, так как многие фторорганические производные обладают весьма ценными свойствами. Необходим фтор и для получения соединений инертных газов.
Наиболее интересными с общехимической точки зрения производными фтора являются фториды инертных газов. Лучше других изученные соединения ксенона могут быть получены из элементов при нагревании, под действием электрического разряда или ультрафиолетовых лучей. Фториды ксенона — ХеF2, ХеF4 и ХеF6 — представляют собой бесцветные легко возгоняющиеся кристаллические вещества
Интересно, что средняя энергия связи Хе-F в них практически одинакова (132,9ё127,9 кДж/моль). Они хорошо (XeF2, ХеF6) или умеренно (ХеF4) растворимы в жидком фтористом водороде, а по донорной способности располагаются в ряд: ХеF42 6. Водой фториды ксенона разлагаются. В процессе гидролиза обычно возникает желтая окраска, которая затем исчезает.
К с е н о д и ф т о р и д медленно образуется под действием дневного света на смесь Хе и F2 уже при обычных условиях (теплота образования 176 кДж/моль). Он обладает характерным тошнотворным запахом. Его давление пара составляет около 3 мм рт. ст. при обычных условиях и 760 мл рт. ст. при 155 °С. Теплота возгонки (сопровождающейся реакцией по схеме 2 ХеF2 = Хе + ХеF4) равна 29,4 кДж/моль. Молекула ХеF2, линейна, связи Хе-F в ней характеризуются длиной 198 пм. По-видимому, они имеют сильно выраженный полярный характер. Для возможности образования этих связей необходимо возбуждение атома ксенона от его нормального нуль валентного состояния (5s25р6) до одного из ближайших двухвалентных, что требует значительной затраты энергии (803 кДж/моль при возбуждении до 5s25p56s1, 924 кДж/моль — до 5s25р56р1 или 953 кДж/моль — до 5s25р55d1). Растворимость ХеF2 в воде составляет около 0,15 моль/л при 0 °С. Раствор является сильнейшим окислителем — потенциал системы ХеF2-Хе в кислой среде равен 2,2 в. Саморазложение раствора по схеме
2 ХеF2 + 2 Н2О = 4 НF + 2 Хе + O2
в кислой среде идет медленно, а в щелочной очень быстро.
К с е н о н т е т р а ф т о р и д образуется из элементов с довольно значительным выделением тепла (251 кДж/моль) и является наиболее устойчивым из всех фторидов ксенона. Молекула его имеет структуру квадрата с атомом Хе в центре, а связь Хе-F характеризуется длиной 195 пм (в кристалле) или 185 пм (в газе) и полярностью. Давление пара составляет около 3 мм рт. ст. при обычных условиях и 760 мм рт. ст. при 146 °С, а теплота возгонки равна 15,3 кДж/моль. Ксенонтетрафторид образует с ХеF2 кристаллический аддукт ХeF2·ХеF4, но не взаимодействует с КF или ВF3. Ртуть он фторирует:
ХеF4 + 2 Нg = 2 НgF2 + Хе,
а раствор его в НF подобным же образом фторирует платину
ХеF4 + Рt = РtF4 +Хе
Иодистый калий (в растворе) количественно реагирует по уравнению:
4 KI + ХеF4 = 4 КF + 2 I2 + Хе
что находит аналитическое использование. Под действием воды ХеF4 разлагается по схеме 3 ХеIV = Хе0 + 2 ХеVI (в кислой среде) или 2 ХеIV = Хе0 + ХеVIII (в щелочной среде).
Был описан также о к с о ф т о р и д ХеОF2, образующийся (в качестве незначительной примеси) при нагревании сильно разбавленной кислородом или воздухом смеси Хе с F2. Для него даются следующие константы: точка плавления 90 °С и точка кипения — около 115 °С. Предполагается, что тот же состав имеет сконденсированный при -80 °С ярко-желтый продукт гидролиза ХеF4 водяным паром. Сообщалось, что это же вещество образовывалось, по-видимому, в результате взаимодействия Хе с большим избытком F2О2 при -118 °С. Однако существование ОХеF2 пока нельзя считать окончательно установленным.
Бесцветный к с е н о н г е к с а ф т о р и д известен в трех различных кристаллических модификациях. Он плавится при 49 °С в желтую жидкость с низкой диэлектрической проницаемостью (e = 4,1 при 55 °С), по-видимому, содержащую тетрамерные ассоциаты. При затвердевании ХеF6 вновь обесцвечивается. Давление его пара (имеющего бледно-желтую окраску) составляет 30 мм рт. ст. при 25 °С и 760 мм рт. ст. при 76 °С. Ксенонгексафторид чрезвычайно химически активен и способен разлагаться со взрывом. Строение его молекулы пока точно не установлено, но известно, что он не обладает обычной для соединений типа ЭF6 симметрией правильного октаэдра. Среднее расстояние d(ХеF) = 190 пм.
Растворение ХеF6 в жидком фтористом водороде сопровождается частичной электролитической диссоциацией по схеме:
ХеF6 + НF = ХеF5++ НF2-
Насыщенный при обычных условиях раствор имеет состав, приблизительно отвечающий формуле ХеF6·6НF. В отличие от тетрафторида ХеF6 образует твердые продукты присоединения и с ВF3, и с фторидами щелочных металлов. Бесцветный Nа2ХеF8 разлагается ниже 100 °С, но Сs2ХеF8 — лишь выше 400 °С. Гораздо менее устойчивы соли типа МХеF7. Так, желтый СsХеF7 переходит в кремовый Сs2ХеF8 уже при 50 °С. Все эти соли чрезвычайно химически активны и бурно реагируют с водой (причем Хe сохраняется в растворе, по-видимому, как ХеО3).
Под действием влажного воздуха ксенонгексафторид частично гидролизуется с образованием о к с о ф т о р и д а ОХеF4. Последний представляет собой бесцветную жидкость (т. пл. -46, т. кип. 102 °С), менее реакционноспособную, чем ХeF6. Она смешивается с жидким фтористым водородом, а с фторидами тяжелых щелочных металлов образует следующие соединения: 3КF·ХеОF4, 3RbF·2ХеОF4, СsF·ХеОF4. Молекула ОХеF4 имеет m = 0,65 и структуру квадратной пирамиды с атомом Хe около середины основания из четырех атомов фтора [d(ХеО) = 1,70, d(ХеF) = 1,90 А, РОХеF = 92°].
Дальнейший медленный гидролиз ХеОF4 (или гидролиз ХеF4 в кислой среде с дисмутацией по схеме 3 Хе+4® Хе0+ 2 Хе+6) ведет к образованию к с е н о н т р и о к с и д a, который может быть выделен в виде крайне взрывчатых бесцветных кристаллов, расплывающихся на воздухе. Теплота образования ХеОз из элементов равна — 401 кДж/моль. В сухом состоянии это сильно эндотермичное соединение способно распадаться со взрывом, но при медленном нагревании выше 40 °С разложение на Хе и O2, идет спокойно (заканчиваясь при 140 °С). Молекула ХеОз имеет форму тригональной пирамиды с атомом Хе в вершине [d(ХеО) = 1,76 А, РОХеО = 103°]. Для средней энергии связи ХеО дается значение 117 кДж/моль.
Взаимодействием ХеО3 с ХеОF4 был получен ХеО2F2. Этот оксофторид представляет собой бесцветные кристаллы (т. пл. 31 °С). Во влажном воздухе он гидролизуется до ХeО3, а в сухом медленно разлагается на ХеF2 и O2. От ХеО3 производятся молекулярные соединения типа МF·ХеО3 (где М — Сs, Rb, К), а также СsС1·ХеО3 и СsВr·ХеО3. Строение их молекул отвечает, по-видимому, формуле М[ХеО3Г]. Фториды термически устойчивы до 200 °С.
Ксенонтриоксид хорошо растворим в воде, но лишь слабо взаимодействует с ней: равновесие по схеме
Н2О + ХеО3Ы Н2ХеО4Ы Н+ + НХеО4-
сильно смещено влево. При рН > 10,5 оно смещается вправо с образованием солей типа МНХеO4 или МН5ХеО6, (где M — Nаё Сs). Отвечающая этим к с е н а т а м кислота была получена при 0 °С взаимодействием ксенонтетрафторида с разбавленным раствором гидрооксида кальция по суммарному уравнению:
3 ХеF4 + 6 Са(ОН)2 = 6 СаF2Ї + Хе + 2 Н2ХеО6
При низких температурах (порядка -25 °С) она может сохраняться длительное время. Ее бариевая соль — Ва3ХеО6 — малорастворима в воде (0,25 г/л при 25 °С) и испытывает термический распад лишь при 125 °С. В сильнощелочной среде шестивалентный ксенон неустойчив (дисмутирует по схеме 4 Хе+6 = Хе0 + 3 Хе+8). Напротив, кислые водные растворы ХеО3 вполне устойчивы. Для окислительных потенциалов системы Хе+6-Хе0даются значения +2,1 в (в кислой среде) и +1,2 в (в щелочной среде).
При действии озона на раствор ХеО3 в 1 М NаОН образуется Nа4ХеО6. Анион этого п е р к с е н а т а имеет структуру слегка искаженного октаэдра со средним расстоянием d(ХеО) = 185 пм. Тетранатрийперксенат может быть выделен в виде бесцветного кристаллогидрата с 6 или 8 Н2О, который обезвоживается около 100 °С, а бурно разлагается лишь при 360 °С. Соль эта малорастворима в воде (растворимость около 0,025 М), но сильно гидролизуется, давая щелочную реакцию. Последнее обусловлено относительной слабостью ксеноновой кислоты, которой отвечают следующие значения последовательных констант диссоциации: К1 = 10-2, К2 = 10-6 и К3 = 3·10-11. Содержащие Хе+8 водные растворы постепенно отщепляют кислород, переходя в растворы Хе+6, причем скорость такого перехода возрастает с уменьшением рН среды (уже при рН = 7 он осуществляется почти мгновенно). Для окислительных потенциалов системы Хе+8-Хе+6 даются значения +2,3 в (в кислой среде) и +0,9 в (в щелочной среде). Смешанным производным этих валентностей является полученное озонированием смеси растворов ХеО3 и КОН взрывчатое желтое молекулярное соединение состава К4ХеО6·2ХеО3.
Взаимодействием Nа4ХеО4 с безводной Н2SO4 при низких температурах был получен желтый к с е н о н т е т р о к с и д (теплота образования из элементов -644 кДж/моль). Молекула ХеО4 имеет структуру тетраэдра с атомом ксенона в центре, а связь ХеО характеризуется ядерным расстоянием d(ХеО) = 174 пм и энергией 88 кДж/моль. Давление пара этого оксида составляет 3 мм рт. ст. при -35 °С. В твердом состоянии он уже ниже 0 °С медленно разлагается на Хе и О2, а в газообразном при комнатной температуре — на ХеО3, Хе и О2.
Сообщалось также об образовании при взаимодействии Nа4ХеО6 и ХеF6 очень летучего ХеО3F2, но выделен он не был.
Имеются отдельные указания на возможность образования в тех или иных условиях нестойкого соединения ксенона с хлором — ХеС12 или ХеС14. Однако все такие указания даются лишь предположительно, и считать, что хлориды ксенона существуют, пока нельзя.
Взаимодействие с фтором радона идет легче, чем ксенона (но состав фторидов не устанавливался), а криптона — гораздо труднее. Известен только к р и п т о н д и ф т о р и д, который был впервые получен действием электроразряда на смесь элементов при -188 °С. Он представляет собой бесцветные кристаллы, давление пара над которыми равно 30 мм рт. ст. при 0 °С, а теплота возгонки составляет 36,8 кДж/моль. Молекула КrF2 линейна, а связь КrF характеризуется ядерным расстоянием d(KrF) = 188 пм, энергией 50 кДж/моль. При низких температурах КrF2 может сохраняться неделями, а при 20 °С за час разлагается около 10 % исходного количества. Его насыщенный раствор в жидком фтористом водороде по составу приблизительно отвечает формуле КrF2·3НF. Получить какие-либо производные аргона (и еще более легких инертных газов) пока не удалось.
Как видно из изложенного выше, сведения о впервые полученных в 1962 г. соединениях инертных газов еще довольно отрывочны (и отчасти недостоверны). Однако сам факт существования этих соединений имеет большое принципиальное значение, так как наиболее наглядно и убедительно опровергает постулат незыблемости электронного октета. Тем самым ставится также вопрос о целесообразности отказа от уже не вполне отвечающего существу названия “инертные газы” (подходящей его заменой могло бы служить название а э р о ф и л ы). О широком практическом использовании соединений инертных газов говорить еще рано, но, например, устойчивый при обычных температурах ХеF4 мог бы служить, удобной реакционной формой фтора (не загрязненного никакими другими химически активными элементами). Следует лишь иметь в виду возможную взрывоопасность этого соединения (из-за образования взрывчатого ХеО3 во влажном воздухе).
В отличие от свободного фтора ф т о р и с т ы й в о д о р о д (НF) и многие его производные используются уже с давних пор.
Непосредственное соединение фтора с водородом сопровождается значительным выделением тепла:
Н2 + F2 = 2 НF + 543 кДж
Реакция протекает обычно со взрывом, который происходит даже при сильном охлаждении газов и в темноте. Практического значения для получения НF этот прямой синтез не имеет, но, в принципе, он может быть использован для создания реактивной тяги.
Промышленное получение фтористого водорода основано на взаимодействии СаF2 с концентрированной Н2SO4 по реакции:
СаF2 + Н2SO4 = СаSO4 +2 НF
Процесс проводят в стальных печах при 120-300 °С. Части установки, служащие для поглощения НF, делаются из свинца.
В качестве реактивного топлива смесь фтора с водородом способна давать удельный импульс 410 сек. Бесцветное пламя, возникающее при взаимодействии этих газов, может иметь температуру до 4500 °С. В лабораторных условиях для получения чистого фтористого водорода применяются обычно небольшие установки изготовленные целиком из платины (или меди). Исходным веществом служит тщательно высушенный бифторид калия (КF·НF), при нагревании разлагающийcя c отщеплением НF. Полученный продукт часто содержит примесь механически увлеченного бифторида. Для очистки его подвергают перегонке при 35-40 °С. Совершенно безводный или близкий к этому состоянию фтористый водород почти мгновенно обугливает фильтровальную бумагу. Этой пробой иногда пользуются для контроля степени его обезвоживания. Более точно такой контроль осуществляется определением электропроводности у безводного фтористого водорода она ничтожно мала, но даже следы воды (как и многих других примесей) резко ее повышают.
Фтористый водород (гидрофторид) представляет собой бесцветную, подвижную и легколетучую жидкость (т. кип. +19,5 °С), смешивающуюся с водой в любых соотношениях. Он обладает резким запахом, дымит на воздухе (вследствие образования с парами воды мелких капелек раствора) и сильно разъедает стенки дыхательных путей. Многие неорганические соединения хорошо растворимы в жидком НF, причем растворы являются, как правило, проводниками и электрического тока.
Связь Н-F характеризуется ядерным расстоянием 0,92 А. По отношению к нагреванию фтористый водород очень устойчив: его термическая диссоциация становится заметной лишь около 3500 °С.
Молекула НF весьма полярна (m = 1,74). С наличием на атомах значительных эффективных зарядов хорошо согласуется резко выраженная склонность фтороводорода к а с с о ц и а ц и и путем образования водородных связей по схеме ···Н-F···Н-F···.
Энергия такой связи составляет около 33,4 кДж/моль, т. е. она прочнее, чем водородная связь между молекулами воды.
Как показывает определение плотности пара, вблизи точки кипения молекулы газообразного фтористого водорода имеют средний состав, приблизительно выражаемый формулой (НF)4. При дальнейшем нагревании ассоциированные агрегаты постепенно распадаются и кажущийся (средний) молекулярный вес уменьшается, причем лишь около 90 °С достигает значения 20, соответствующего простой молекуле НF.
Критическая температура фтористого водорода равна 188 °С, критическое давление 64 атм. Теплота испарения жидкого НF в точке кипения составляет лишь 7,5 кДж/моль. Столь низкое значение (примерно в 6 раз меньшее, чем у воды при 20 °С) обусловлено тем, что само по себе испарение мало меняет характер ассоциации фтористого водорода (в отличие от воды).
Подобно плотности (0,99 г/см3), диэлектрическая проницаемость жидкого фтористого водорода (84 при 0 °С) очень близка к значению ее для воды. Существующая у жидкого фтористого водорода ничтожная электропроводность обусловлена его незначительной ионизацией по схеме:
НF + НF + НF Ы Н2F+ + НF2-
связанной с характерной для НF склонностью к образованию иона г и д р о д и ф т о р и д а — НF2- [имеющего линейную структуру с атомом водорода в центре и d(FF) = 227 пм]. Напротив, образование иона ф т о р о н и я (Н2F+) для НF нехарактерно, что и ограничивает самоионизацию (К = 2·10-11). Тенденция к образованию иона НF2-,, накладывает свой отпечаток на всю химию фтористого водорода.
Помимо воды, из неорганических соединений в жидком HF хорошо растворимы фториды, нитраты и сульфаты одновалентных металлов (и аммония), хуже — аналогичные соли Mg, Сa, Sr и Вa. По рядам Li-Сs и Мg-Ва, т. е. по мере усиления металлического характера элемента, растворимость повышается. Щелочные и щелочноземельные соли других галоидов растворяются в НF с выделением соответствующего галоидоводорода. Соли тяжелых металлов в жидком HF, как правило, нерастворимы. Наиболее интересным исключением является Т1F, растворимость которого очень велика (в весовом отношении около 6: 1 при 12 °С). Практически нерастворимы в жидком НF другие галоидоводороды. Концентрированная серная кислота взаимодействует с ним по схеме:
Н2SO4 + 3 НF Ы Н3О++ НSO3F + НF2-
Жидкий фтористый водород является лучшим из всех известных растворителем белков.
Растворы воды и солей в жидком фтористом водороде хорошо проводят электрический ток, что обусловлено диссоциацией, например, по схемам:
Н2О + 2 НF Ы Н3О++ НF2-
КNО3 + 2 НF Ы НNО3 + К+ + НF2-
НNО3 + 4 НF Ы Н3О+ + NО2+ + 2 НF2-
Аналогичное отношение к НF характерно и для многих кислородсодержащих органических молекул. Так, в водной среде глюкоза является типичным неэлектролитом, а в жидком НF наоборот, типичным электролитом за счет взаимодействия по схеме:
С6Н12O6 + 2 НF Ы [С6Н12О6·Н]+ + НF2-
Кристаллы твердого фтористого водорода слагаются из зигзагообразных цепей ···Н-F···Н-F···Н-F···, образованных при посредстве водородных связей. Расстояние d(FF) в таких цепях — 249 пм, а угол зигзага — 120°. Теплота плавления твердого НF (т. пл. -83 °С, плотность 1,6 г/см3) составляет 3,8 кДж/моль, что близко к значению для льда. Для жидкого фтористого водорода наиболее вероятно одновременное существование и цепей, и колец из молекул НF.
Химическая активность НF существенно зависит ~н отсутствия или наличия воды. Сухой фтористый водород не действует на большинство металлов. Не реагирует он и с оксидами металлов. Однако если реакция с оксидом начнется хотя бы в ничтожной степени, то дальше она некоторое время идет с самоускорением, так как в результате взаимодействия по схеме
МО + 2 НF = МF2 + Н2О
количество воды увеличивается.
Случаи взаимодействия сухого фтористого водорода с оксидами металлов и металлоидов, рассмотренные выше, могут служить типичным примером аутокаталитических реакций, т. е. таких процессов, при которых катализатор (в данном случае — вода) не вводится в систему извне, а является одним из продуктов реакции.
Как показывает рис. Ч11-4, скорость подобных процессов сначала, по мере увеличения в системе количества катализатора, нарастает до некоторого максимума, после чего начинает уменьшаться вследствие понижения концентраций реагирующих веществ.
***********РИС
Подобным же образом действует фтористый водород и на окислы некоторых металлоидов. Практически важно его взаимодействие с двуокисью кремния — SiO2 (песок, кварц), которая входит в состав стекла. Реакция идет по схеме
SiO2 + 4 НF = SiF4 + 2 Н2O
Поэтому фтористый водород нельзя получать и сохранять в стеклянных сосудах.
На взаимодействии НF и SiO2 основано применение фтористого водорода для травления стекла. При этом вследствие удаления частичек SiO2 его поверхность становится матовой, что и используют для нанесения на стекло различных меток, надписей и т. п.
Перед фигурным травлением стекла его обычно покрывают тонким слоем воска, а затем снимают этот слой на тех местах, которые должны быть протравлены. Под действием паров НF места эти становятся матовыми, тогда как под действием плавиковой кислоты они остаются прозрачными. Матовое травление в жидкости достигается предварительным добавлением к плавиковой кислоте нескольких процентов фтористого аммония.
В водном растворе НF ведет себя как одноосновная кислота средней силы. Продажный раствор этой фтористоводородной (иначе, п л а в и к о в о й) кислоты содержит обычно 40% НF.
Техническая плавиковая кислота обычно содержит ряд примесей — Fе, Рb, Аs, Н2SiF6, SO2) и др. Для грубой очистки ее подвергают перегонке в аппаратуре, изготовленной целиком из платины (или свинца), отбрасывая первые порции дистиллята. Если этой очистки недостаточно, то техническую кислоту переводят в бифторид калия, затем разлагают его нагреванием и растворяют получающийся фтористый водород в дистиллированной воде. Крепкая плавиковая кислота (более 60% НF) может сохраняться и транспортироваться в стальных емкостях. Для хранения плавиковой кислоты и работы с ней в лабораторных условиях наиболее удобны сосуды из некоторых органических пластмасс. Крупным потребителем фтористоводородной кислоты является алюминиевая промышленность.
Растворение фтористого водорода в воде сопровождается довольно значительным выделением тепла (59 кДж/моль). Характерно для него образование содержащей 38,3 % НF и кипящей при 112 °С азеотропной смеси (по другим данным 37,5 % и т. кип. 109 °С). Такая азеотропная смесь получается в конечном счете при перегонке как крепкой, так и разбавленной кислоты.
При низких температурах фтористый водород образует нестойкие соединения с водой состава Н2О·НF, Н2О·2НF и Н2О·4НF. Наиболее устойчиво из них первое (т. пл. -35 °С), которое следует рассматривать как фторид оксония — [Н3O]F.
Помимо обычной электролитической диссоциации по уравнению
HF Ы H+ + F- (К = 7·10-4),
для растворов фтористоводородной кислоты характерно равновесие:
F- + НF Ы НF2’
Значение константы этого равновесия ([НF2’]/[F’][НF]=5) показывает, что в не очень разбавленных растворах НF2’ содержится больше анионов чем простых анионов F’. Например, для приводимых ниже общих нормальностей (С) приближенно имеем:
С [НF] [Н+'] [F-] [HF2’]
0,100 0,088 (88 %) 0,009 (9 %) 0,006 (6 %) 0,003 (3 %)
1,000 0,890 (89 %) 0,006 (6 %) 0,010 (1 %) 0,050 (5 %)
Фтористоводородная кислота (ацидофторид) более или менее энергично реагирует с большинством металлов. Однако во многих случаях реакция протекает лишь на поверхности, после чего металл оказывается защищенным от дальнейшего действия кислоты слоем образовавшейся труднорастворимой соли. Так ведет себя, в частности, свинец, что и позволяет пользоваться им для изготовления частей аппаратуры, устойчивой к действию НF.
Соли фтористоводородной кислоты носят название ф т о р и с т ы х или ф т о р и д о в. Большинство их малорастворимо в воде — из производных наиболее обычных металлов хорошо растворяются лишь фториды Nа, К, Ag, A1, Sn и Нg. Все соли плавиковой кислоты ядовиты. Сама она при попадании на кожу вызывает образование болезненных и трудно заживающих ожогов (особенно под ногтями). Поэтому работать с плавиковой кислотой следует в резиновых перчатках.
Весьма характерно для фтористого водорода образование продуктов присоединения к фторидам наиболее активных металлов. Соединения эти, как правило, хорошо кристаллизуются и плавятся без разложения. Примером могут служить производные калия — КF·НF (т. пл. 239 °С), КF·2НF (62 °С), КF·3НF (66 °С) и КF·4НF (72 °С). Строение этих продуктов присоединения отвечает, вероятно, формулам вида К[F(НF)n] с водородными связями между ионом F- и молекулами HF. Разбавленные растворы гидродифторида калия (КНF2) применяются иногда для удаления пятен от ржавчины.
Работа с фтористым водородом и другими фторидами требует соблюдения мер предосторожности, так как все соединения фтора ядовиты. Сам фтористый водород, помимо резкого раздражения слизистых оболочек, вызывает также разрушение ногтей и зубов. Кроме того, он способствует осаждению кальция в тканях. Средство первой помощи при острых отравлениях фторидами служит 2 %-ный раствор СаС12. При ожогах плавиковой кислотой пораженное место следует длительно (несколько часов) промывать струей холодной воды, а затем наложить на него компресс из свежеприготовленной 20 %-ной взвеси MgО в глицерине.
Хроническое отравление фторидами может быть вызвано как повышенным их содержанием в питьевой воде, так и вдыханием их с воздухом в виде пыли. В результате подобного отравления наблюдается разрушение зубной эмали. Существенно увеличивается также хрупкость костей, что создает предпосылки для их переломов. Имеются указания на то, что повышенное содержание фторидов в воде и воздухе способствует заболеванию зобом. Помимо фторной промышленности, с возможностью хронического отравления фтористыми соединениями приходится особенно считаться при выработке алюминия и суперфосфата. Предельно допустимой концентрацией связанного фтора в воздухе производственных помещений считается 5·10-4 мг/л.
Практическое применение НF довольно разнообразно. Безводный используется главным образом при органических синтезах, а плавиковая кислота — для получения фторидов, травления стекла, удаления песка с металлического лития, при анализах минералов и т. д. Широкое применение находят также некоторые фториды которые будут рассмотрены при соответствующих элементах.
2. Хлор.
По распространенности в природе хлор близок к фтору на его долю приходится 0,02 % от общего числа атомов земной коры. Человеческий организм содержит 0,25 вес. % хлора.
Природный хлор состоит из смеси двух изотопов — 35Сl (75,5 %) и 37Сl (24,5 %). Он был впервые получен (действием МnО2 на соляную кислоту ) в 1774 г., но установление его элементарной природы последовало лишь в 1810 г.
Подобно фтору, основная масса хлора поступила на земную поверхность из горячих недр Земли. Даже в настоящее время с вулканическими газами ежегодно выделяются миллионы тонн и НСl и НF. Еще гораздо более значительным было такое выделение в минувшие эпохи.
Первичная форма нахождения хлора на земной поверхности отвечает его чрезвычайному распылению. В результате работы воды, на протяжении многих миллионов лет разрушавшей горные породы и вымывавшей из них все растворимые составные части, соединения хлора скапливались в морях. Усыхания последних привело к образованию во многих местах земного шара мощных залежей NаС1, который и служит исходным сырьем для получения соединений хлора.
Будучи наиболее практически важным из всех галоидов, хлор в громадных количествах используется для беления тканей и бумажной массы, обеззараживания питьевой воды (примерно 1,5 г на 1 м3) и в других отраслях техники. Ежегодное мировое потребление хлора исчисляется миллионами тонн.
Рис. Ч11 —5. Принципиальная схема электролизера для полу —
Основным промышленным методом получения хлора является электролиз концентрированного раствора NаС1. Принципиальная схема электролизера показана на рис. VII-5 (А - аноды, Б - диафрагма, В - катод). При электролизе на аноде выделяется хлор (2С1-- 2е- = С12), а в при катодном пространстве выделяется водород (2Н+ + 2е- = Н2) образуется NаОН.
При практическом осуществлении электролиза раствора NaCl расход электроэнергии на получение 1 т хлора составляет около 2700 кВт·ч. Полученный хлор под давлением сгущается в желтую жидкость уже при обычных температурах. Хранят и перевозят его в стальных баллонах, где он заключен под давлением около 6 атм. Баллоны эти должны иметь окраску защитного цвета с зеленой поперечной полосой в верхней части.
Для лабораторного получения хлора обычно пользуются действием MnO2 или КМnO4 на соляную кислоту:
МnО2 + 4 НСl = МnСl2 + Сl2 + 2 Н2О
2 КМnO4 + 16 НCl = 2 КCl + 2 МnСl2 + 5 Сl2 + 8 Н2О
Вторая реакция протекает значительно энергичнее первой (требующей подогревания).
Свободный хлор представляет собой желто-зеленый газ, состоящий из двухатомных молекул. Под обычным давлением он сжижается при -34 °С и затвердевает при -101 °С. Один объем воды растворяет около двух объемов хлора. Образующийся желтоватый раствор часто называют «хлорной водой».
Критическая температура хлора равна 144 °С, критическое давление 76 атм. При температуре кипения жидкий хлор имеет плотность 1,6 г/см3, а теплота его испарения составляет 20,5 кДж/моль. Твердый хлор имеет плотность 2,0 г/см3 и теплоту плавления 6,3 кДж/моль. Кристаллы его образованы отдельными молекулами С12 (кратчайшее расстояние между которыми равно 334 пм).
Связь Сl-Сl характеризуется ядерным расстоянием 198 пм. Термическая диссоциация молекулярного хлора по уравнению С12 + 242 кДж Ы 2 С1 становится заметной примерно с 1000 °С.
Атом хлора имеет в основном состоянии структуру внешнего электронного слоя 3s23р5 и одновалентен. Возбуждение его до ближайшего трехковалентного уровня 3s23р44s1 требует затраты 857 кДж/моль.
Энергия присоединения электрона к нейтральному атому хлора оценивается в 355 кДж/моль. Сродство к электрону хлора (аналогично и других галоидов) может быть вычислено при помощи рассмотрения реакций образования хлористых солей по отдельным стадиям. Например, для NаС1 имеем:
1) Nа (т) = Nа (г) — 109 кДж (теплота возгонки)
2) 1/2 С12 (г) = С1 (г) — 121 кДж (теплота диссоциации)
3) Na (г) = Nа+(г) + е- — 493 кДж (энергия ионизации)
4) С1(г) + е- = Сl-(г) + Х кДж (искомое сродство к электрону)
5) Nа+(г) + Сl-(г) = NаС1(т) +777 кДж (энергия кристаллической решетки)
в сумме: Nа(т) + 1/2 С12(г) = NаСl(т) + (Х+777-493-121-109) кДж
С другой стороны, непосредственно определенная на опыте теплота образования NаС1 из элементов равна: Nа(т) + 1/2 С12(г) = NаС1(т) + 410 кДж. Следовательно, по закону Гесса, Х + 777 - 493 - 121 - 109 = 410, откуда Х = 356 кДж.
Ион С1- — характеризуется эффективным радиусом 181 пм и энергией гидратации 351 кДж/моль. Для ковалентного радиуса хлора принимается половина ядерного расстояния молекулы С12, т. е. 99 пм.
Растворимость хлора в воде меняется с температурой следующим образом:
Температура, °С | 10 | 15 | 20 | 25 | 30 | 40 | 50 | 60 | |
Растворимость V на 1V H2O | 4,6 | 3,1 | 2,7 | 2,3 | 2,0 | 1,8 | 1,4 | 1,2 | 1,0 |
Описаны два кристаллогидрата хлора — С12·6Н2О и С12·8Н2О. В действительности они могут иметь переменный состав, так как являются клатратами.
Значительно хуже (примерно в 4 раза), чем в воде, растворяется хлор в насыщенном растворе NаС1, которым поэтому и удобно пользоваться при собирании хлора над жидкостью. Наиболее пригодным для работ с ним органическим растворителем является четыреххлористый углерод (СС14), один объем которого растворяет при обычных условиях около 50 объемов хлора.
Основным потребителями хлора являются органическая технология (получение хлорированных полупродуктов синтеза) и целлюлозно-бумажная промышленность (отбелка). Значительно меньше потребляется хлор в неорганической технологии, санитарной технике и других областях. Интересно недавно предложенное использование хлора для обработки металлов: под его действием с достаточно нагретой (инфракрасным излучением) поверхности все шероховатости удаляются в форме летучих хлоридов. Такой метод химической шлифовки особенно применим к изделиям сложного профиля. Было показано также, что струя хлора легко прорезает достаточно нагретые листы из жаростойких сплавов.
Хлор обладает резким запахом. Вдыхание его вызывает воспаление дыхательных путей. В качестве средства первой помощи при острых отравлениях хлором применяется вдыхание паров смеси спирта с эфиром. Полезно также вдыхание паров нашатырного спирта.
Предельно допустимой концентрацией свободного хлора в воздухе производственных помещений считается 0,001 мг/л. Пребывание в атмосфере, содержащей 0,01% хлора и выше, быстро ведет к тяжелому заболеванию. Признаком острого отравления является появление мучительного кашля. Пострадавшему необходимо прежде всего обеспечить полный покой; полезно также вдыхание кислорода.
По своей характерной химической функции хлор подобен фтору — он также является о д н о в а л е н т н ы м м е т а л л о и д о м. Однако активность его меньше, чем у фтора. Поэтому последний способен вытеснять хлор из соединений.
Тем не менее химическая активность хлора очень велика —
он соединяется почти со всеми металлами (иногда лишь в присутствии следов воды или при нагревании) и со всеми металлоидными элементами, кроме С, N и O. Важно отметить, что при полном отсутствии влаги хлор не действует на железо. Это и позволяет хранить его в стальных баллонах.
Взаимодействие хлора с фтором при нагревании смеси сухих газов происходит лишь выше 270 °С. В этих условиях с выделением тепла (50 кДж/моль) образуется бесцветный хлорфторид — С1F (т. пл. -156, т. кип. -100 °C). Газообразный С1F обладает сильным своеобразным запахом (отличным от запахов хлора и фтора).
Взаимодействием хлорфторида с фторидами Сs, Rb и К под высоким давлением были получены бесцветные малостойкие соли типа МС1F2, содержащие в своем составе линейный анион С1F2-. При нагревании они экзотермически разлагаются около 250 °С.
Нагреванием С1F с избытком фтора может быть получен бледно-зеленоватый трехфтористый хлор (хлортрифторид) — СlF3 (т. пл. -76, т. кип. +12 °С). Соединение это также экзотермично (теплота образования из элементов 159 кДж/моль) и по запаху похоже на С1F. Молекула С1F3 полярна (m = 0,55) и имеет показанную на рис. У11 —7 плоскую структуру.
Последняя производится от тригональной бипирамиды, у которой два направления треугольного основания закрываются свободными электронными парами атома хлора. Критическая температура С1F3 равна 154 °С, плотность в жидком состоянии 1,8 г/см3, теплота испарения 27,6 кДж/моль. Вблизи точки кипения пар трехфтористого хлора несколько ассоциирован по схеме: 2 С1F3Ы (С1F3)2 + 12,5 кДж. Для димера вероятна мостиковая структура (по типу F2С1F2С1F2).
Жидкий С1F3 смешивается с жидким НF в любых соотношениях, причем имеет место слабое взаимодействие по схеме: НF + С1F3Ы НС1F4 + 16,7 кДж. Образующийся ацидохлортетрафторид не выделен, но производящиеся от него соли типа МС1F4, (где М — Сs, Rb, К) известны. По-видимому, они могут быть получены не только прямым сочетанием МF и С1F3, но и фторированием соответствующих хлоридов (3000 атм, 300 °С).
Нагреванием смеси С1F3 с избытком фтора под высоким давлением может быть получен бесцветный хлорпентафторид — С1F5 (т. пл. -93, т. пл. -13 °С). Теплота его образования из элементов 251 кДж/моль. Молекула С1F5, имеет строение квадратной пирамиды из атомов фтора, вблизи основания которой располагается атом хлора. В отсутствие влаги этот газ при обычных условиях устойчив, а водой разлагается. Он является энергичным фторирующим агентом, но корродирует металлы слабее, чем С1F3.
Фториды хлора характеризуются исключительной реакционной способностью. Например, в парах С1F3 стеклянная вата самовоспламеняется. Почти столь же энергично взаимодействуют с ним и такие сами по себе чрезвычайно устойчивые вещества, как MgО, СаО, А12O3 и т. п. Так как С1F3 сжижается при обычных температурах уже под небольшим давлением и легко отщепляет фтор, его удобно использовать для транспортировки фтора. Помимо различных реакций фторирования, отмечалась возможность применения этого вещества как окислителя реактивных топлив и зажигательного средства в военной технике. По трифториду хлора имеется обзорная статья.
Взаимодействие хлора с в о д о р о д о м по реакции:
Н2 + С12 = 2 НС1 + 184 кДж
при обычных условиях протекает крайне медленно, но нагревание смеси газов или ее сильное освещение (прямым солнечным светом, горящим магнием и т. д.) сопровождается взрывом.
Детальное изучение этой реакции позволило выяснить сущность ее отдельных стадий. Прежде всего за счет энергии (hn) ультрафиолетовых лучей (или нагревания) молекула хлора диссоциирует на атомы, которые затем реагируют с молекулами водорода, образуя НСl и атом водорода. Последний в свою очередь реагирует с молекулой хлора, образуя НС1 и атом хлора, и т. д.:
1) С12 + hn = С1 + С1 (первоначальное возбуждение)
2)… С1 + Н2 = НС1 + Н
3)… Н+ С12 = НС1 + С1 и т. д.
Таким образом, получается как бы цепь последовательных реакций, причем за счет каждой первоначально возбужденной молекулы Сl2 образуется в среднем 100 тыс. молекул НС1. Реакции подобного типа называются цепными. Они играют важную роль при протекании многих химических процессов.
Фотохимическая диссоциация молекулы хлора на атомы вызывается светом с длиной волны 550 нм. Обеим стадиям цепной реакции образования хлористого водорода соответствуют следующие термохимические уравнения:
С1 + Н2 + 1 кДж = НС1 + Н и Н + С12 = НС1 + С1 + 188 кДж. Энергия активации первой из этих реакций составляет 25, а второй 8 кДж/моль. Малыми значениями этих энергий и обусловлено быстрое развитие цепи.
Очевидно, что цепь могла бы оборваться, если бы протекала реакция: Н + С1 = НС1. Такая возможность не исключена, однако вероятность осуществления этой реакции очень мала, так как концентрация атомов ничтожна по сравнению с концентрацией молекул и поэтому несравненно больше шансов имеет столкновение каждого из атомов с молекулой другого элемента, чем обоих атомов друг с другом. С другой стороны, произведенные на основе экспериментальных данных расчеты показывают, что даже при столкновении обоих атомов соединение между ними происходит далеко не всегда,
Рис 1-2 888888888
наоборот, процент успешных встреч очень мал. По этим же причинам цепи редко обрываются в результате реакций: С1+ Сl = С12 и Н + Н = Н2. Так, последняя из них осуществляется в газовой фазе лишь при одном столкновении из каждого миллиона.
«Огромное большинство реакций при ближайшем рассмотрении являются цепными реакциями» (Н. Н. Семенов). Это нередко вызывает отклонение их действительной молекулярности от отвечающей простейшему суммарному уравнению. В частности наблюдаемая на опыте бимолекулярность реакции образования волы из элементов обусловлена именно ее цепным характером: начало цепи дает (с энергией активации 188 кДж/моль) реакция Н2 + О2 = 2 ОН, после чего цепь разветвляется по схемам: ОН + Н2 = Н2О + Н, Н + О2 = ОН + О, О + Н2 = ОН + Н и т. д. Как видно из этих схем, число активных участников реакции (ОН, Н, О) последовательно возрастает, вследствие чего процесс протекает с самоускорением. Это и характерно для разветвленных цепных реакций, в отличие от неразветвленных, примером которых может служить синтез хлористого водорода.
Большие количества НС1 получают в технике как побочный продукт хлорирования органических соединений по схеме
RН + Cl2 = RС1 + НС1
где R — органический радикал. Однако для получения чистой соляной кислоты основное значение имеет прямой синтез. Исходным сырьем служат при этом хлор и водород, одновременно выделяющийся при электролизе раствора NаС1. Спокойное протекание процесса обеспечивается смешиванием обоих газов лишь в момент взаимодействия.
Еще один метод промышленного получения НС1 основан на взаимодействии NаС1 и концентрированной Н2SO4 по реакциям
NаС1 + Н2SO4 = NаНSO4 + НС1
NаС1 + NаНSO4 = Nа2SO4 + НС1
Первая из них протекает в значительной степени уже при обычных условиях и практически нацело — при слабом нагревании; вторая осуществляется лишь при более высоких температурах. Для проведения процесса служат специальные механизированные печи большой производительности.
Максимальная температура водородно-хлорного пламени составляет около 2200 °С. Для технического синтеза НС1 служит установка, схематически показанная на рис. Ч11 —8. После первоначального поджигания смесь хлора с водородом продолжает гореть спокойным пламенем, образуя хлористый водород. Последний проходит затем сквозь две поглотительные башни с водой, в которых и образуется соляная кислота. Используемый в системе принцип противотока, т. е. противоположных направлений движения газа и жидкости, обеспечивает полноту поглощения НС1 и позволяет проводить весь процесс непрерывно.
Основной частью показанной на рис. Ч11—9 механизированной печи для получения НСl является муфель А, со всех сторон обогреваемый горячими газами, идущими из топки Б. Внутри муфеля медленно вращается мешалка, гребенки которой устроены таким образом, что реагирующая масса передвигается ими от центра муфеля (куда подаются исходные вещества) к его краям. Выделяющийся хлористый водород после его обеспыливания и охлаждения улавливается водой, а образующийся Nа2SO4 сбрасывается в бункер Г (откуда грузится на вагонетки). Печь работает непрерывно и перерабатывает за сутки несколько тонн NаС1.
С теоретической стороны интересен метод получения хлористого водорода путем пропускания смеси хлора с водяным паром сквозь слой раскаленного угля. Реакция в этих условиях идет по уравнению:
2 Сl2 + 2 Н2О + С = СO2 + 4 HCl + 280 кДж
Так как она сильно экзотермична, уголь поддерживается в раскаленном состоянии за счет ее тепла. Практически этот метод не применяется (так как получающийся влажный хлористый водород сильно разъедает детали установки).
Хлористый водород (гидрохлорид) представляет собой бесцветный газ. В отсутствие влаги он при обычных температурах не действует на большинство металлов и их оксиды. Газообразный кислород окисляет его только при нагревании.
Молекула НСl характеризуется ядерным расстоянием d(HCl) = 128 пм, энергией связи 431 кДж. Хлористый водород плавится при -114 °С и кипит при -85 °С. Распад НС1 на элементы становится заметным примерно при 1500 °С.
Под давлением около 70 атм хлористый водород сжижается уже при обычных температурах и, подобно хлору, может транспортироваться к местам потребления а стальных баллонах. Жидкий хлористый водород обладает малой диэлектрической проницаемостью (4,6 при обычных температурах) и является плохим растворителем подавляющего большинства неорганических соединений. Растворимы в нем, например, хлориды олова и фосфора. Интересно, что РF3 растворим и жидком НС1, но не взаимодействует с ним, тогда как АsF3 и SbF3 испытывают полный сольволиз по схеме
ЭF3 + 3 НС1 = 3 НF + ЭС13
С темно-красным окрашиванием растворяется иод. Жидкий НС1 смешивается с жидкими СО2 и Н2S.
Предельно допустимой концентрацией хлористого водорода в воздухе производственных помещений считается 0,005 мг/л. Наличие уже 0,05 мг/л быстро вызывает раздражение в носу и гортани, колотье в груди, хрипоту и ощущение удушья. При хроническом отравлении малыми концентрациями НС1 особенно страдают зубы, эмаль которых подвергается быстрому разрушению.
Реакция в газовой фазе по уравнению
О2 + 4 НС1 = 2 Н2О + 2 С12 + 117 кДж
обратима. Ниже 600 °С равновесие ее смещено вправо, выше 600 °С — влево. На этой реакции был основан часто применявшийся ранее метод технического получения хлора: пропусканием смеси НС1 с воздухом над нагретым до 450 °С катализатором (пропитанный раствором СuС12 асбест) удавалось получать хлор с выходом около 70 % от теоретического. В связи с характерной для последнего времени дефицитностью хлора подобный метод может вновь приобрести промышленное значение.
На воздухе хлористый водород дымит вследствие образования с парами воды капелек тумана. Растворимость его весьма велика: при обычных условиях 1 объем воды способен поглотить около 450 объемов хлористого водорода.
Раствор НCl в воде называется хлористоводородной (иначе со л я н о й) кислотой. Она относится к числу наиболее сильных кислот. Реактивная соляная кислота обычно имеет плотность 1,19 г/см3 и содержит около 37 % хлористого водорода. Состав ее близок к формуле HCl·3,5H2O.
Растворимость НС1 в воде меняется с температурой следующим образом:
Температура, °С | 10 | 15 | 20 | 25 | 30 | 40 | 50 | 60 | |
Растворимость в V на 1 V Н2О | 507 | 474 | 459 | 442 | 426 | 412 | 386 | 362 | 339 |
Растворение сопровождается выделением тепла (до 75 кДж/моль НСl). Давление хлористого водорода над крепкой соляной кислотой при 20 °С приводится ниже:
| Концентрация НСl, % | 24 | 26 | 28 | 30 | 32 | 34 | 36 | 38 |
| P, мм рт. ст. | 1,0 | 2,2 | 4,9 | 10,6 | 23,5 | 50,5 | 105 | 210 |
При смешивании концентрированной НС1 со снегом происходит резкое понижение температуры. Содержащий 25 вес.% НС1 водный раствор замерзает лишь при — 86 °С.
Органические жидкости поглощают хлористый водород гораздо хуже воды. Например, при обычных условиях эфир растворяет НС1 примерно в 3,5 раза, а бензол — в 50 раз меньше, чем вода.
Хлористый водород образует с водой азеотропную смесь, которая кипит под обычным давлением при 109 °С и содержит 20,2 % НС1.
Охлаждением концентрированных водных растворов хлористого водорода могут быть выделены кристаллогидраты НСl с 6, 3, 2 и 1 молекулами Н2О, плавящиеся с разложением соответственно при -70, -25, -18, -15 °С. Последний из них по структуре является хлоридом оксония (Н3О+С1-), в кристаллогидрате НСl·2Н2О четко выявляются катионы Н5О2+ с очень короткой водородной связью [d(OO) = 241 пм] между двумя молекулами воды, а структура тригидрата соответствует формуле Н5О2+Сl-·Н2О. С жидким хлором хлористый водород дает молекулярные соединения состава С12·2НС1 и С12·НС1, плавящиеся соответственно при -121 и -115 °С.
Техническая соляная кислота выпускается крепостью не менее 31% НС1 (синтетическая) или 27 % HСl (из NаСl). Приблизительное процентное содержание НС1 в водном растворе легко найти, умножив на 2 число дробных долей его плотности. Например, при плотности 1,19 г/см3 процентное содержание получается равным 19·2 = 38 %. Следовательно, и обратно, зная процентное содержание НС1 в соляной кислоте той или иной крепости, можно приближенно оценить ее плотность. Путем приготовления 1,184 н. раствора НС1 удобно создавать среду с рН = 0 (при 25 °С). Как видно из приводимых ниже приблизительных данных, в крепких водных растворах (с моляльностью больше двух) коэффициент активности (f) хлористого водорода значительно превышает единицу:
m | 1 | 2 | 4 | 6 | 8 | 10 | 12 | 14 |
| f | 0,8 | 1 | 2 | 3 | 6 | 10 | 17 | 27 |
Соляная кислота очень сильно разъедает многие металлы. Транспортируют ее в стеклянных бутылях или гуммированных (т. е. покрытых слоем резины) металлических емкостях. Гуммирование может быть заменено введением в кислоту специальных добавок — ингибиторов.
Соляная кислота содержится в желудочном соке (около 0,3 %) и играет важную роль, так как способствует перевариванию пищи и убивает различные болезнетворные бактерии (холеры, тифа и др.). Если последние попадают в желудок вместе с большим количеством воды, то вследствие разбавления раствора НСl они выживают и вызывают заболевание организма. Поэтому во время эпидемий особенно опасна сырая вода. При повышении концентрации НС1 в желудке ощущается «изжога», которую устраняют, принимая внутрь небольшое количество NаНСО3 или МgО. Наоборот, при недостаточной кислотности желудочного сока соляная кислота прописывается для приема внутрь (по 5-15 капель 8,3 %-ной НСl на 1/2 стакана воды до или во время еды).
Подобно другим сильным кислотам, НС1 энергично взаимодействует со многими металлами, оксидами металлов и т. д. Соли ее называются х л о р и с т ы м и или х л о р и д а м и. Большинство их хорошо растворимо в воде. Из производных наиболее обычных металлов труднорастворимы хлориды серебра и свинца. Ежегодное мировое потребление соляной кислоты исчисляется миллионами тонн. Широкое практическое применение находят также многие ее соли.
Длительное взаимодействие безводных СsС1 и НС1 при -78 °С ведет к образованию нестойкого СsС1·НС1 (давление пара > 400 мм рт. ст. при 30 °С). Хлориды других элементарных катионов подобных соединений не образуют, но было получено аналогичное производное катиона [N(СН3)4]+ и показано, что ион НСl2- аналогичен иону HF2-. В отличие от НF, образование такого иона [d(СlСl) = 314 пм] для HCl не характерно.
Непосредственное взаимодействие хлора с кислородом не приводит к образованию кислородных соединений хлора. Они могут быть получены лишь косвенными методами. Для рассмотрения путей их образования целесообразно исходить из обратимой реакции между хлором и водой:
С12 + Н2О + 25 кДж Ы НС1 + НОС1
При обычных условиях в насыщенном растворе гидролизовано около трети всего растворенного хлора.
Взаимодействие хлора с перекисью водорода первоначально протекает по уравнению:
С12 + Н2О2 = 2 НОС1
Однако избытком Н2О2 хлорноватистая кислота восстанавливается:
НОС1 + Н2О2 = НС1 + Н2О + О2
Из образующихся при гидролизе хлора двух кислот — соляной и хлорноватистой (HOС1) — первая является очень сильной, а вторая — очень слабой (слабее угольной). Это резкое различие в силе обеих кислот можно использовать для их разделения.
Если в растворе взболтать порошок мела (СаСО3) и затем пропускать в него хлор, то образующаяся соляная кислота реагирует с мелом по уравнению:
CaCO3 + 2 НС1 = СаС12 + СО2 + Н2О
а хлорноватистая накапливается в растворе. Подвергая реакционную смесь перегонке получают в приемнике разбавленный раствор НОС1.
Будучи соединением малоустойчивым, НОС1 медленно разлагается даже в таком разбавленном растворе. Соли хлорноватистой кислоты называются г и п о х л о р и т а м и. И сама HС1O, и ее соли являются очень с и л ь н ы м и окислителями.
Наиболее концентрированные растворы НОС1 образуются при взаимодействии жидкого С12О с охлажденной водой (обе жидкости ограниченно растворимы друг в друге). Для получения растворов крепостью до 5 М удобно обрабатывать хлором (без избытка) взвесь оксида ртути в четыреххлористом углероде. Образующаяся в растворе С12О извлекается затем холодной водой. Возможно также получение раствора хлорноватистой кислоты по реакции:
2 С12 + Вi2О3 + Н2О = 2 ВiОСlЇ + 2 НОС1
Молекула НОС1 имеет угловое строение с параметрами d(НО) = 97, d(ОС1) = 169 пм, РНОС1 = 103°.
Хлорноватистая кислота обладает характерным запахом. Ее разбавленные растворы почти бесцветны, а более крепкие имеют желтый цвет. Константа кислотной диссоциации НОС1 при обычных условиях равна 4·10-8. Диссоциация ее по основному типу (т. е. НОСl Ы НО’+ С1•) экспериментально не обнаружена. Однако имеются косвенные указания на ее возможность. Например, с органическими соединениями НОС1 способна реагировать по схемам (R — органический радикал)
RН + НОС1 = RОН + НС1 и RН + НОСl = Н2О + RС1
т. е. и как окислитель, и как хлорирующее вещество.
Принципиальная возможность амфотерной диссоциации НОСl вытекает из общетеоретических соображений. Однако в присутствии С1’ непосредственно обнаружить С1• нельзя (из-за реакции по схеме С1•+ С1’ = С12·аq).
Так как при переходе от НОН к НОС1 отрицательный характер кислорода ослабевает, относительная вероятность внедрения в него протона уменьшается. Поэтому выражаемое схемой
ОН3+ + НОС1 Ы Н2О + Н2ОС1+
(или, учитывая неопределенную гидратированность обоих ионов, Н•+ НОСl Ы Н2О + Сl•) равновесие должно быть сильно смещено влево, но по мере повышения концентрации Н• должно несколько смещаться вправо. Экспериментально доказать возможность основной диссоциации НОС1 можно было бы, вероятно, подвергнув электролизу свежеприготовленный раствор С12О в холодной 30 %-ной серной кислоте: возникающий за счет приведенного выше равновесия положительный ион хлора должен был бы перемещаться к катоду.
Практический метод получения гипохлоритов основан на использовании приводимой выше обратимой реакции взаимодействия хлора с водой. Поскольку оба вещества правой части равенства — НСl и НОCl — дают в растворе ионы Н•, а оба исходных продукта — С12 и Н2О — таких ионов не образуют (точнее, почти не образуют), равновесие можно сместить вправо, связывая ионы Н•.
Добиться этого проще всего добавлением к реакционной смеси какой-нибудь щелочи. Так как по мере своего образования ионы Н• будут связываться ионами ОН’ в недиссоциированные молекулы воды, равновесие практически нацело сместится вправо. Применяя, например, КОН, имеем
С12 + Н2О Ы НОС1 + НС1
НОС1 + НС1 + 2 КОН = КОС1 + КС1 + 2 Н2О
или в общем: С12 + 2 КОН = КОС1 + КС1 + Н2О
В результате взаимодействия хлора с раствором щелочи получается, следовательно, смесь солей хлорноватистой и соляной кислот. Этот процесс имеет большое техническое значение, так как образующийся раствор гипохлорита обладает сильными окислительными свойствами и широко применяется для беления тканей (хлопковых и льняных) и бумаги.
Ввиду слабости хлорноватистой кислоты под действием углекислого газа воздуха происходит частичное ее выделение из раствора гипохлорита:
NаОС1 + CO2 + Н2О Ы NаНСО3 + НОС1
Беление основано на окислении хлорноватистой кислотой различных загрязняющих ткань веществ. Так как наличие NаС1 отбелке не вредит, применяют непосредственно раствор, получающийся в результате реакции хлора со щелочью.
Раствор этот часто называют «жавелевой водой». На текстильных и бумажных фабриках ее иногда получают электролизом раствора NаС1 без диафрагмы. При этом первоначально образуются NаОН и С12, которые, взаимодействуя друг с другом, и дают «жавелевую воду». После беления ею необходимо очень тщательно промывать ткани, так как избыток NаОС1 постепенно разъедает их.
Кристаллический натрийгипохлорит может быть получен отгонкой воды из его раствора под уменьшенным давлением, Выделяется он в виде кристаллогидрата NаОСl·5Н2О (т. пл. 45 °С), который легко переходит в NаОС1·Н2О. Последняя соль малоустойчива, а при нагревании до 70 °С разлагается со взрывом. Значительно устойчивее LiОС1·Н2О, который при обычных условиях выдерживает длительное хранение.
Опыт показывает, что окислительная активность гипохлоритов максимальна при таких значениях рН (близких к 7), когда в растворе одновременно имеются соизмеримые концентрации и ионов ОС1-, и молекул НОС1. Вероятно, это связано с равновесием по схеме:
ОСl- + НОСl Ы ОС1Н + ОСl-
Хотя оно и должно быть сильно смещено влево, его существование все же обеспечивает постоянную возможность временного возникновения неустойчивых молекул и з о х л о р н о в а т и с т о й кислоты, структура которых позволяет предполагать наличие у них повышенной тенденции к отщеплению активного атома кислорода.
При взаимодействии хлора с более дешевой щелочью — Са(ОН)2 («гашеной известью») — образуется х л о р н а я известь. Реакция может быть приближенно выражена уравнением
С12 + Са(ОН)2 = Сl-Са-ОCl + H2O
согласно которому хлорная известь является с м е ш а н н о й солью соляной и хлорноватистой кислот. Она представляет собой белый порошок, обладающий сильными окислительными свойствами, и используется главным образом для дезинфекции.
Формула Са(С1)ОС1 отражает основной состав хлорной (иначе — белильной) извести лишь схематично. Получаемый хлорированием Са(ОН)2 продукт представляет собой смесь различных двойных и тройных соединений, образованных молекулами Са(ОСl)2, Са(ОН)2, СаС12 и кристаллизационной воды.
На воздухе хлорная известь постепенно разлагается, в основном по схеме:
2 Са(С1)ОСl + СО2 = СаС12 + СаСО3 + С12O
При действии на нее соляной кислоты выделяется хлор:
Са(С1)ОС1 + 2 НС1 = СаС12 + Н2О + С12
Этим иногда пользуются для его получения — хлорную известь смешивают с гипсом и из образовавшейся массы формуют кубики, которыми заряжают аппарат для получения газов. Качество хлорной извести оценивают обычно количеством хлора, образующимся при действии на нее соляной кислоты. Хорошие продажные сорта приближенно отвечают составу 3Са(С1)ОСl·Ca(ОН)2·nН2О и содержат около 35 вес.% «активного» (т. е. выделяющегося при действии соляной кислоты) хлора.
Для получения более высокопроцентной хлорной извести, состоящей главным образом из Са(ОС1)2, хлорированию подвергают не сухой Са(ОН)2, а взвесь его в небольшом количестве воды. При 30 °С реакция идет в основном по уравнению
2 Са(ОН)2 + 2 С12 = Са(ОС1)2 + СаС12 + 2 Н2О
причем большая часть образующегося Са(ОС1)2 выделяется в виде мелкокристаллического осадка. Получаемый после отфильтровывания и высушивания технический продукт содержит 45-70 % активного хлора. При взаимодействии с водой он растворяется почти полностью, тогда как обычная хлорная известь дает объемистый осадок Са(ОН)2.
Свободная хлорноватистая кислота испытывает в растворе три различных типа превращений, которые протекают независимо друг от друга. и поэтому называются параллельными реакциями:
1) НОС1 = НС1 + О
2) 2 НОС1 = Н2О + С12О
3) 3 НОС1 = 2 НС1 + НСlO3
Все эти процессы способны протекать одновременно, но их относительные скорости сильно зависят от имеющихся условий. Изменяя последние, можно добиться того, что превращение пойдет практически нацело по какому-нибудь одному направлению.
Под действием прямого солнечного света разложение хлорноватистой кислоты идет по п е р в о м у из них. Так же протекает оно в присутствии веществ, способных легко присоединять кислород, и некоторых катализаторов (например, солей кобальта).
При нагревании крепкого раствора хлорной извести в присутствии солей кобальта распад ее идет по уравнению:
2 Са(С1)ОСl = 2 СаС12 + O2 + 92 кДж
Реакцией этой иногда пользуются для лабораторного получения кислорода.
При распаде по в т о р о м у типу получается газообразный продукт — оксид хлора (С12О). Эта реакция идет в присутствии водоотнимающих веществ (например, СаС12). Оксид хлора представляет собой взрывчатый желто-бурый газ с запахом, похожим на запах хлора. При действии С12О на воду образуется НОС1, т. е. окись хлора является ангидридом хлорноватистой кислоты.
Молекула С12О полярна (m = 0,78) и характеризуется треугольной структурой [d(СlO) = -170 пм, Рa = 111°. Энергия связи О-С1 оценивается в 205 кДж/моль. Оксид хлора (дихлормоноксид) легко сгущается в красно-коричневую жидкость (т. пл. -121, т. кип. +2 °С), которая может длительно сохраняться при -78 °С, но более или менее быстро разлагается при обычных условиях (в основном по схеме 4 С12О = 2 С1О2 + 3 С12). Получать его удобно, действуя при охлаждении хлором на свежеосажденный сухую оксид ртути. Реакция идет по уравнению:
2 НgО + 2 Cl2 = С1НgОНgС1 + С12O + 79 кДж
Взрыв жидкого оксида хлора иногда происходит уже при переливании ее из одного сосуда в другой, а газообразной — при нагревании или соприкосновении со многими способными окисляться веществами. Он протекает по уравнению
2 С12О = 2 С12 + О2 + 150 кДж
Энергия активации этой реакции составляет 105 кДж/моль.
Оксид хлора хорошо растворим в СС14. Еще лучше он растворяется в воде за счет взаимодействия по реакции
Сl2O + Н2О Ы 2 НОС1
равновесие которой сильно смещено вправо (К = [С12О]/[НОС1]2 = 1·10-3 при 0 °С). Охлаждением крепких водных растворов С12О может быть получен кристаллогидрат хлорноватистой кислоты состава НОСl·2H2O (т. пл. -36 °С).
Распад НОС1 по т р е т ь е м у типу особенно легко идет при нагревании. Поэтому действие хлора на горячий раствор щелочи выражается суммарным уравнением
3 С12 + 6 КОН = КС1О3 + 5 КС1 + 3 Н2О
Продуктами реакции являются КС1 и калийная соль хлорноватой кислоты (НС1О3). Так как соль эта малорастворима в холодной воде, при охлаждении раствора она осаждается.
Свободная НС1О3 может существовать только в растворе. Она является с и л ь н о й к и с л о т о й (диссоциированной приблизительно так же, как НС1 и НNО3) и энергичным окислителем. Соответствующий ей ангидрид неизвестен.
В противоположность свободной НС1О3, для ее солей (х л о р а т о в) окислительные свойства в растворах не характерны. Большинство из них бесцветно (как и сама НС1О3) и хорошо растворимо в воде. Все они сильно ядовиты.
Переход гипохлорита в хлорат осуществляется, вероятно, с участием изохлорноватистой кислоты по схемам:
НСlO + СlO- = НСl + СlO2-и НСlO + СlO2 = НСl + СlO3-
Анион СlO3- имеет структуру треугольной пирамиды с хлором в вершине [d(ClO) = 145 пм, РОСlO = 106°].
Из солей хлорноватой кислоты практически наиболее важен КС1О3 (т. пл. 368 °С), который может быть получен электролизом горячего раствора КС1. Хлорат калия применяется в спичечном производстве, при изготовлении сигнальных ракет и т. д. Легкорастворимый в воде NаС1O3 (т. пл. 262 °С) является прекрасным средством для уничтожения сорных трав (на железнодорожном полотне и т. д.).
Энергия активации термического разложения чистого КС1О3 равна 226 кДж/моль (следует учитывать, что процесс этот может протекать со взрывом). Расплавленный КСlO3 энергично поддерживает горение. Смеси его с легко окисляющимися веществами (серой, фосфором, сахаром и др.) взрываются от удара.
Раствор хлорноватой кислоты обычно получают действием серной кислоты на Ba(ClO3)2 ( т. пл. 414 °С). Отфильтровав осадок ВаSO4, можно путем упаривания при низких температурах (в вакууме) сконцентрировать раствор примерно до 40 % содержания НС1О3. Получается густая бесцветная жидкость приблизительного состава НС1О3·7Н2О, при нагревании выше 40 °С разлагающаяся. Такой раствор характеризуется столь сильно выраженными окислительными свойствами, что при соприкосновении с ним бумага, вата и т. п. воспламеняются. Более разбавленные растворы НС1О3 в обычных условиях довольно устойчивы. При сильном охлаждении они становятся густыми и вязкими, но не закристаллизовываются.
При длительном совместном нагревании фторидов и хлоритов некоторых двухвалентных металлов в присутствии уксусной кислоты происходит взаимодействие по схеме
МF2 + M(С1O3)2 = 2 МС1O3F
с образованием соответствующей соли фторохлорноватой кислоты (Н2С1О3F). Таким путем синтезировались хорошо растворимые ф т о р х л о р а т ы ряда лвухвалентных металлов (например, Сu(ClО3F·5H2O). Под действием на их растворы иона Са — осадок СаF2, начинает медленно выделяться лишь при кипячении, т. е. ион С1О3F” оказывается довольно устойчивым по отношению к гидролизу. Были получены также некоторые другие производные фторхлорноватой кислоты.
Осторожным восстановлением хлоратов может быть получен диоксид хлора (С1О2). Он представляет собой взрывчатый желтый газ, обладающий сильно выраженными окислительными свойствами.
В лабораторных условиях СlO2 удобно получать по реакции 2 КСlO3 + H2С2О4 = К2СО3 + СО2 + Н2О + 2 СlO2
нагреванием до 60 °С увлажненной смеси КСlO3 и щавелевой кислоты (Н2С2O4). Другим удобным методом лабораторного получения СlO2 является проводимая при 90 °С с тщательно осушенным хлором реакция по уравнению:
Сl2 + 2 АgСlO3 = 2 АgСl + 2 СlO2 + O2
При охлаждении ниже +10 °С диоксид хлора сгущается в красно-коричневую жидкость и может быть таким путем отделен от углекислого газа или кислорода.
Молекула С1О2 полярна (m = 1,78) и характеризуется треугольной структурой [d(СlO) = 147 пм, Рa = 118°]. Энергия связи С1-О равна 251 кДж/моль.
В твердом состоянии диоксид хлора (хлордиоксид) представляет собой желтовато-красные кристаллы (т. пл. -59 °С). Плотность ее пара отвечает простой формуле, но для раствора в СС14 установлено наличие частичной димеризации по схеме 2 С1O2Ы С12O4 (константа равновесия равна 0,18 при 25 °С). Запах ClO2 одновременно похож на запах хлора и азотной кислоты. Он начинает ощущаться при 0,002 %-ном содержании С1О2 в воздухе. В темноте чистый диоксид хлора устойчив по на свету или при наличии даже следов хлоридов постепенно разлагается. Будучи эндотермичным (теплота образования — 105 кДж/моль) и малоустойчивым соединением, С1О2 может взрываться при нагревании или соприкосновении со способными окисляться веществами.
Диоксид хлора хорошо растворим в воде (20: 1 по объему при 4 °С) с желто-оранжевой окраской жидкости. Разбавленные растворы (до 8 г/л) в темноте устойчивы но на свету медленно разлагаются (с образованием НСlO3 и НС1). Известен кристаллогидрат С1О2·6Н2О.
Используется С1О2 главным образом для отбелки или стерилизации различных материалов (бумажной массы, муки и др.). Установлено, что с его помощью можно производить обесфеноливание сточных вод химических заводов.
В связи с быстрым ростом потребления С1О2 для технических целей, был предложен ряд методов его промышленного получения. Примером может служить метод, основанный на экзотермической реакции
2 NаClO3 + SO2 + Н2SO4 = 2 NаНSO4 + 2 ClО2
проводимой с приблизительно 4 М серной кислотой (содержащей значительную примесь хлорид-иона).
Исходя из С1О2 довольно сложным путем было получено устойчивое -78 °С, но начинающее разлагаться уже при -45 °С темно-коричневое твердое вещество, состав которого отвечает формуле С12О3. Является ли оно действительно оксидом трехвалентного хлора (или представляет собой смесь других его оксидов), пока не ясно.
При медленном пропускании тока фтора под поверхность охлажденной до -50 °С диоксида хлора происходит ее фторирование с образованием фторхлордиоксида (FClO2). Вещество это представляет собой бесцветный газ (т. пл. -115, т. кип. -6 °С), довольно устойчивый по отношению к нагреванию, но весьма гигроскопичный. Гидролиз его идет по схеме:
FСlO2 + Н2О = НF + НСlO3
Взаимодействие FСlO2 с НС1 (при -110 °С) протекает по уравнению:
2 FСlO2 + 2 НСl = 2 НF + Сl2 + 2 СlO2
т. е. СlСlO2 оказывается совершенно неустойчивым. Вместе с тем были получены некоторые солеобразные производные СlO2+, например СlO2SbF6 (т. пл. 235 °С).
Взаимодействие С1О2 с раствором КОН медленно протекает по уравнению
2 С1О2 + 2 КОН = КС1О3 + КС1О2 + Н2О
с образованием солей двух кислот — хлорноватой и хлористой. Сама хлористая кислота (НС1О2) малоустойчива. По силе и окислительной активности она промежуточна между НОС1 и НС1О3. Соли ее (х л о р и т ы) используются при отбелке тканей.
Хлористую кислоту (К = 1·10-2) можно получить по реакциям:
ВаО2 + 2 С1О2 = Ва(С1О2)2 + О2 и
Ва(С1О2)2 + Н2SO4 = ВаSO4Ї +2 НС1O2
Она известна только в разбавленных растворах, при хранении которых очень быстро разлагается, в основном, по схеме:
4 HСlO2 = 2 СlO2 + НСlO3 + НCl + Н2О
Ион СlO2, имеет треугольную структуру [d(СlO) = 155 пм, РОСlO = 111°]. Хлориты, как правило, бесцветны и хорошо растворимы в воде [за исключением желтых АgСlO2 (1,7 г/л) и Рb(СlO2)2 (0,35 г/л при 0 °С)]. В отличие от гипохлоритов, они характеризуются наличием сильно выраженных окислительных свойств только в кислой среде. С другой стороны, под действием КМnO4 хлориты способны окисляться до хлоратов. Имеются указания на возможность образования некоторых хлоритов при непосредственном взаимодействии соответствующего металла (например, Ni) с раствором СlO2. В твердом состоянии многие соли НСlO2 легко взрываются при нагревании или ударе.
Наиболее практически важным хлоритом является NаСlO2. Эту соль удобно получать по реакции:
2 СlO2 + РbО + 2 NаОН = РbО2Ї + 2 NаСlO2 + Н2О
Выше 100 °С разлагается в основном по схеме:
3 NаСlO2 = 2 NаСlO3 + NаС1
При нагревании КС1О3 плавится, а около 400 °С начинает разлагаться, причем распад может идти по двум основным направлениям:
1) 4 КС1О3 = 4 КС1 + 6 О2 + 180 кДж
2) 4 КС1О3 = КС1 + 3 КС1О4 + 171 кДж
Реакция протекает преимущественно по первому типу при наличии катализатора (МnО2 и т. п.), по второму — в его отсутствие. Образующийся при распаде по второму типу х л о р а т калия) очень малорастворим в воде и поэтому легко отделяется от хорошо растворимого хлористого калия.
Действием на калийперхлорат концентрированной серной кислоты может быть получена свободная хлорная кислота (НС1О4), представляющая собой бесцветную, сильно дымящую на воздухе жидкость:
КС1О4 + Н2SO4Ы КНSO4 + НС1O4
Так как под уменьшенным давлением НС1O4 перегоняется без разложения, ее легко выделить из реакционной смеси.
Безводная НСlO4 малоустойчива и иногда взрывается просто при хранении, но ее водные растворы вполне устойчивы. Как о к и с л и т е л ь HClO4 гораздо менее активна, чем НС1O3, и в разбавленных растворах практически не обнаруживает окислительных свойств. Напротив, к и с л о т н ы е свойства выражены у нее исключительно резко: по-видимому, она является одной из самых сильных кислот.
Соли НСlO4, за немногими исключениями
(рис. Ч11 —6),
легко растворимы в воде. Многие из них хорошо растворяются также в органических растворителях (спирте и т. п.). Подобно самой кислоте, большинство перхлоратов бесцветно.
Калийперхлорат применяется для приготовления некоторых взрывчатых веществ. При 610 °С он плавится и одновременно начинает разлагаться, в основном по уравнению:
KСlO4 = КСl + 2 O2
Получают КСlO4 обычно электролизом раствора КСlO3. Реакция идет по схеме:
КСlO3 + Н2О = Н2 (катод) + КСlO4 (анод).
При перегонке разбавленных растворов НСlO4 сначала отгоняется вода, затем разбавленная кислота и, наконец, при 203 °С начинает перегоняться азеотропная смесь, содержащая 72 % HСlO4 (близкая к составу НСlO4·2Н2О и замерзающая лишь при -18 °C). Так как кипение последней сопровождается частичным разложением, перегонку HClO4 лучше проводить под уменьшенным давлением (при 20 мм рт. ст. азеотропная смесь перегоняется около 111 °С). Концентрированная (72%) кислота дымит на воздухе и весьма гигроскопична, но устойчива при хранении и не разлагается под действием света. Промышленностью обычно выпускается 30-70 %-ная НСlO4.
Молекула HСlO4 имеет форму пирамиды с тремя атомами кислорода в основании [d(СlO) = 141 пм], гидроксильной группой в вершине [d(С10) = 164 пм] и углом О-Сl=O, равным 106°. Безводная хлорная кислота (т. пл. — 101, т. кип. +16'С при 18 мм рт. ст.) представляет собой весьма подвижную жидкость, тогда как ее крепкие водные растворы имеют маслянистую консистенцию. Их охлаждением может быть получен плавящийся лишь при +50 °С кристаллогидрат НСlO4·Н2О, который следует рассматривать как перхлорат оксония — [Н3О]СlO4. Частичное образование последнего по схеме
3 НСlO4Ы [Н3О]СlO4 + Сl2O7 + 12,5 кДж
(с константой равновесия К = 1·10-4) имеет место и в безводной хлорной кислоте. Именно этой реакцией (в силу последующего распада Сl2O7 по схеме 2 Сl2O7 = 4 СlO2 + 3 O2 + 117 кДж) обусловлена, вероятно, неустойчивость безводной хлорной кислоты. Очень сильные взрывы может вызвать ее соприкосновение со способными окисляться веществами. Хлорная кислота находит применение при анализах, в частности для выделения более летучих кислот из их солей.
В разбавленных водных растворах НСlO4 не восстанавливается такими сильными восстановителями, как НI, Н2S, SO2 и водород в момент выделения. Даже концентрированная кислота становится очень активным окислителем лишь при температуре кипения (когда она легко растворяет, в частности, специальные стали).
Хотя НСlO4 является одной из самых сильных из кислот, наличие недиссоциированных молекул в ее растворах установлено несколькими методами. Как видно из рис. ЧП —10, заметным оно становится лишь в достаточно крепких растворах. Для константы равновесия НСlO4Ы Н+ + СlO4’ получено значение К = 38. По другим данным, хлорная кислота ионизирована в растворах еще значительнее, чем то показано на рис. VII-10.
Входящий в состав перхлоратов анион СlO4- представляет собой тетраэдр с хлором в центре [d(СlO) = 144 пм].
Из безводных перхлоратов без разложения плавится только LiСlO4 (т. пл. 236 °С).
Вообще говоря, их термическое разложение может идти по двум схемам: с образованием хлорида металла и кислорода или оксида металла, хлора и кислорода. Для солей Сs, Rb, К характерен первый путь, для солей Nа, Li, Ва, Sr, Сa преимущественно он же, а для солей Мg и большинства других металлов основным становится второй путь распада.
Растворимость некоторых перхлоратов (г на 100 г растворителя при 25 °С) в воде, спирте и ацетоне сопоставлена ниже:
LiClO4 | NaClO4 | KClO4 | Mg(ClO4)2 | Ca(ClO4)2 | Ba(ClO4)2 | |
H2O | 60 | 210 | 2,1 | 100 | 189 | 198 |
C2H5OH | 152 | 15 | 0,01 | 24 | 166 | 125 |
(CH3)2CO | 137 | 52 | 0,16 | 43 | 150 | 125 |
Безводный перхлорат лития хорошо растворим и в эфире (с образованием 6 М раствора), тогда как кристаллогидрат LiСlO4·3Н2О растворим весьма мало. Следует отметить, что растворы перхлоратов в органических жидкостях, как правило, взрывоопасны. Некоторые перхлораты (особенно NН4СlO4) используются в реактивной технике.
Взаимодействием 72 %-ной НСlO4 с фтором получен бесцветный фторперхлорат — FСlO4. Это малоустойчивое соединение (т. пл. -167, т. кип. -10 °С) обладает резким запахом и весьма реакционноспособно. И в газообразном, и в жидком состоянии оно может разлагаться со взрывом.
Длительным взаимодействием избытка СsСlO4 с ClSO3F при -45 °С был получен хлорперхлорат СlСlO4. Вещество это описывается как устойчивая лишь при низких температурах светло-желтая жидкость (т. пл. -117 °С). Наличие в молекуле хлорперхлората положительно поляризованного атома хлора устанавливается протекающими при -78 °С реакциями по схемам
НCl + СlOСlO3 = Сl2 + НСlO4 и
АgСl + СlOСlO3 = Сl2 + АgСlO4
Взрывоопасность СlСlO4 меньше, чем FСlO4.
Если фторперхлорат является продуктом замещения на фтор в о л о р о д а хлорной кислоты, то в качестве продукта аналогичного замещения ее г и д р о к с и л а можно рассматривать фторхлортриоксид («перхлорилфторид») — FСlO3. Последний образуется при действии фтора на сухой КСlO3 и представляет собой бесцветный газ (т. пл. -148, т. кип. -47 °С) с характерным сладковатым запахом. Удобнее получать его по схеме:
МСlO4 + НSО3F = МНSО4 + FСlO3
действием на перхлорат смеси хлорсульфоновой кислоты и SbF5 (которая играет роль катализатора). Теплота образования FСlO3, из элементов равна — 21 кДж/моль, а для энергий связей даются значения 251 (FСl) и 238 (СlO) кДж/моль. Молекула FСlO3 имеет структуру несколько искаженного тетраэдра с хлором около центра [d(СlO) = 140, d(FСl) = 161 А, РОС1O = 115°, РFСlO = 103°] и практически неполярна (m = 0,02).
Фторхлортриоксид термически устойчив до 400 °С, не гидролизуется даже горячей водой (и холодными щелочами), нерастворим в жидком фтористом водороде, умеренно токсичен и сам по себе невзрывчат (но способен давать взрывчатые смеси с некоторыми органическими веществами). Так как его критическая температура довольно высока (+95 °С), он может храниться и транспортироваться в сжиженном состоянии (при 25 °С давление пара составляет 12 атм). Окислительная активность FСlO3 в обычных условиях невелика, но быстро возрастает при нагревании. Поэтому реакции окисления им хорошо поддаются температурному регулированию. Вещество это представляет значительный интерес для реактивной техники. Существует также указание на то, что оно обладает наивысшим из всех газов значением диэлектрической проницаемости.
При слабом нагревании под уменьшенным давлением смеси безводной НС1О4 с фосфорным ангидридом (Р2О5) отгоняется бесцветная маслянистая жидкость, которая представляет собой хлорный ангидрид, образующийся по реакции
2 НСlO4 + Р2О5 = 2 НРО3 + Сl2O7
От сильного нагревания (и удара) Сl2O7 взрывается, однако он все же устойчивее, чем Сl2O и СlO2. При взаимодействии его с водой медленно образуется хлорная кислота.
Хлорный ангидрид (т. пл. -93, т. кип. 83 °С) является сильно эндотермичным соединением (теплота образования из элементов -251 кДж/моль). Строение его молекулы отвечает формуле О2-Сl-O-СlO3. Угол при кислородном атоме, соединяющем обе пирамиды СlO3 составляет 119° [при d(ОСl) = 171 пм], а угол O- Сl=О равен 115° [d(СlO) = 141 пм]. Молекула характеризуется отчетливо выраженной полярностью (m = 0,72). С такими веществами, как сера, фосфор, бумага, опилки и т. п., Сl2O7; при обычных температурах не реагирует, но соприкосновение его с иодом сопровождается взрывом. Хлорный ангидрид смешивается с четыреххлористым углеродом в любых соотношениях. При термическом разложении Сl2O7 первичным актом является разрыв одной из связей О-С1 (с образованием радикалов СlO3 и СlO4). Энергия этой связи оценивается в 201 кДж/моль.
Из двух радикалов, первично возникающих при термическом распаде хлорного ангидрида более или менее устойчивому существованию способен, по-видимому, лишь ClO3. Триоксид хлора (хлортриоксид) образуется при действии на СlO2 озона и представляет собой темно-красное масло (т. замерз. +3 °С). Жидкость примерно на 99 % состоит из удвоенных молекул (Сl2O6), тогда как в парообразном состоянии равновесие Сl2O6 + 8 кДж Ы 2 СlO3 очень сильно смещено вправо.
Хотя выше уже приводились названия кислородных кислот хлора и их солей, однако полезно сопоставить эти названия:
Кислота Формула Название солей
Хлорноватистая НОС1 г и п о х л о р и т ы
Хлористая НС1O2 х л о р и т ы
Хлорноватая НС1O3 х л о р а т ы
Хлорная НС1О4 п е р х л о р а т ы
Структурные формулы всех четырех кислот приводятся ниже:
Н-O-С1 Н-O-С1=O Н-O-С1=O O
ЅЅ ЅЅ
O H-O-Cl=O
ЅЅ
O
Как видно из этих формул, валентность хлора в рассматриваемых кислотах меняется по ряду: +1, +3, +5, +7.
Если сопоставить друг с другом кислородные кислоты хлора по важнейшим для них химическим свойствам — кислотности и окислительной активности, — получается следующая схема:
усиление кислотных свойств
——————————————®
НОС1 НС1О2 НС1О3 НС1О4
¬——————————————
увеличение окислительной активности
Кислотность изменяется, следовательно, противоположно окислительной активности. Последняя, в общем, тем больше, чем кислота менее устойчива. Действительно, хлорноватистая и хлористая кислоты более или менее устойчивы только в разбавленных растворах, концентрацию хлорноватой можно довести уже до 40 %, тогда как хлорная известна в безводном состоянии. Первые три кислоты в растворах постепенно разлагаются, а хлорная может сохраняться сколь угодно долго. Соответствующие соли обычно значительно устойчивее свободных кислот, но относительная их устойчивость примерно такова же.
Так как наиболее устойчивой из всех кислородных кислот хлора является НСlO4, можно было бы ожидать, что при взаимодействии хлора со щелочью должны сразу образовываться ее соли. Однако сперва получаются менее устойчивые соединения, которые затем лишь постепенно (быстрее — при нагревании) переходят в более устойчивые. На основе изучения ряда подобных случаев уже Гей-Люссак (1842 г.) наметил так называемое п р а в и л о с т у п е н е й р е а к ц и и: при химических процессах вначале обычно образуются не наиболее устойчивые вещества, а самые близкие по неустойчивости к исходной системе.
Во всех тех случаях, когда дальнейшие превращения относительно менее устойчивых продуктов реакции осуществляются очень быстро или, наоборот, очень медленно, мы практически их либо не замечаем, либо не считаем промежуточными продуктами. Поэтому выражаемое правилом ступеней реакции обобщение сразу бросается в глаза. Между тем при рассмотрении хода протекания химических процессов оно часто оказывается весьма полезным.
4. Подгруппа брома.
Содержание в земной коре брома составляет 3·10-5 %, а иода 4 ·10-6 %. По характеру распределения в природе оба элемента очень похожи на хлор, но образование вторичных скоплений для них нехарактерно. Содержание в природе астата ничтожно мало, и свойства этого элемента почти не изучены.
Природный бром состоит из смеси изотопов 79Вr (50,5 %) 81Br (49,5 %), тогда как иод является “чистым” элементом — состоит из атомов 127I. Для астата известны только радиоактивные изотопы с небольшой продолжительностью жизни атомов (в среднем 12 ч для наиболее долгоживущего 210At).
Иод был открыт в 1811 г., бром — в 1826 г. Существование астата предсказывалось уже Д. И. Менделеевым. Элемент этот был получен искусственно в 1940 г. Происхождение брома и иода земной поверхности такое же как хлора и фтора — основные массы обоих элементов выделялись из горячих недр Земли в форме своих водородных соединений.
Основными источниками промышленного получения брома являются воды некоторых соляных озер (0,01-0,5 % Вr) и морская вода (в среднем 0,007 % Вr). Частично он добывается также из бромистых соединений, примеси которых обычно содержатся в природных месторождениях калийных солей, и из буровых вод нефтеносных районов (0,01-0,1 % Br).
Для промышленной добычи иода основное значение имеют именно буровые воды, содержащие в среднем 0,003%. Другим источником этого элемента является зола морских водорослей.
Для получения свободных брома и иода можно воспользоваться вытеснением их хлором. Бром выделяется из раствора исходной соли в виде тяжелой жидкости, иод — в твердом состоянии.
При получении брома из морской (или озерной) воды ее подкисляют серной кислотой до рН = 3,5 и обрабатывают хлором. Выделяющийся бром перегоняют током воздуха в раствор соды, который после достаточного насыщения бромом подкисляют. Реакции протекают по уравнениям:
2 NаВr + Сl2 = 2 NаСl + Вr2, затем
3 Вr2 + 3 Nа2СО3 = 5 NаВr + NаВrО3 + 3 СО2 и, наконец,
5 NаВr + NаВrO3 + 3 Н2SO4 = 3 Na2SO4 + 3 Вr2 + 3 Н2О.
Технический бром часто содержит примесь хлора. Для очистки его обрабатывают концентрированным раствором СаВr2, причем хлор вытесняет бром, который при разбавлении раствора выделяется в виде тяжелого слоя, содержащего лишь очень немного (порядка 0,05 %) растворенной воды.
В безводном состоянии бром может быть получен отгонкой из смеси с концентрированной Н2SO4. Тройной точке на его диаграмме состояния отвечает температура -7,3 °С и давление 46 мм рт. ст. Жидкий бром имеет весьма низкое значение диэлектрической проницаемости (e = 3). Охлаждение его насыщенного водного раствора ведет к образованию кристаллогидрата Вr2·8Н2О (т. пл. 6 °С). Известен также нестойкий кристаллосольват с бензолом состава Вr2·С6Н6 (т. пл. -14 °С).
Так как содержание иода в буровых водах очень мало, основной задачей при получении является его концентрирование. Это обычно достигается выделением иода в свободном состоянии, чаще всего — по реакции:
2 NаI + 2 NаNО2 + 2 Н2SO4 = 2 Na2SO4 + I2 + 2 NО + 2 Н2О
с последующей его адсорбцией на активированном угле. Из последнего иод извлекают горячим раствором едкого натра по реакции:
3 I2 + 6 NаOH = 5 NаI + NаIO3 + 3 Н2О
После насыщения раствора подкислением его вновь выделяют свободный иод по реакции
5 NаI + NаIO3 + 3 Н2SO4 = 3 Nа2SO4 + 3 I2 + 3 Н2О
Морская вода содержит около 5·10-6 % иода, который извлекается из нее некоторыми водорослями и накапливается ими. Например, широко используемая населением Китая и Японии в качестве пищевого продукта ламинария (морская капуста) содержит в воздушно-сухом состоянии около 0,5 % иода.
Для получения иода из золы морских водорослей ее обрабатывают водой и после упаривания раствора оставляют его кристаллизоваться. Бульшая часть содержащихся в золе хлористых и сернокислых солей выпадает при этом в осадок, а иодистые соли, как более растворимые, остаются в растворе. Иод извлекают затем обработкой раствора хлором (или МnО2 и Н2SO4).
По основным физическим свойствам бром и иод закономерно укладываются в один ряд с хлором и фтором, как это видно из приводимой ниже таблицы (в которую включен также водород):
| При обычных условиях | |||||
| Химическая формула | Молекулярный вес округленно | Агрегатное состояние | Цвет | Тпл°С | Ткип°С |
H2 | 2 | Газ | Бесцветный | -259 | -253 |
F2 | 38 | Газ | Почти Бесцветный | -220 | -188 |
Cl2 | 71 | Газ | Желто-зеленый | -101 | -34 |
Br2 | 160 | Жидкость | Темно-коричневый | -7 | 59 |
I2 | 254 | Твердое вещество | Темно-серый | 114 | 186 |
Плотность брома равна 3,1, иода 4,9 г/см3. Так как давление пара твердого иода очень велико, он при нагревании легко возгоняется. Возгонкой технического иода пользуются для его очистки.
Для температур плавления и кипения астата даются значения 227 и 317 °С. Теплоты плавления брома, иода и астата равны соответственно 10,5, 15,9 и 20,9 кДж/моль, а теплоты их испарения (при температурах кипения) — 29,7, 41,8 и 54,3 кДж/моль. Критическая температура брома равна 311, иода — 553 °С. Интересно, что давление паров брома и иода в присутствии индифферентных газов (N2 и др.) выше, чем при той же температуре без них.
Тройной точке на диаграмме состояния иода соответствует температура 116 °С и давление 90 мм рт. ст. Для получения жидкого иода необходимо, следовательно, создать такие условия, чтобы парциальное давление его паров превышало 90 мм рт. ст. Это проще всего достигается нагреванием достигается нагреванием большого количества кристаллов иода в колбе с узким горлом.
Жидкий иод имеет довольно высокое значение диэлектрической проницаемости (e = 11). Он растворяет S, Sе, Те, иодиды ряда металлов и многих органические соединения. Раствор в нем иодистого калия проводит электрический ток. Сам иод диссоциирован по схеме I2Ы I- + 1+, но диссоциация эта очень мала: [I+][I-] = 10-42.
Темно-фиолетовые пары иода и красно-коричневые пары брома (в еще большей степени) обладают резким запахом. По действию на организмы бром близок к хлору. Бром применяется главным образом для выработки специальных добавок к моторным бензинам. Иод в виде 5 %-ного спиртового раствора («иодной настойки») применяется для стерилизации ран. Соединения обоих тяжелых галогенов имеют большое значение для фотографии, медицины и т. д. Ежегодная мировая выработка брома исчисляется десятками тысяч тонн, иода — тысячами тонн.
Физиологическая роль б р о м и с т ы х соединений в нормальной жизнедеятельности организма еще недостаточно выяснена. К их дополнительному введению наиболее чувствительна центральная нервная система: бромиды используются в медицине как успокаивающие средства при повышенной возбудимости. Чрезмерное их накопление способствует появлению кожных сыпей. Выводятся они из организма очень медленно (главным образом, с мочой). По токсическому действию паров бром похож на хлор. При ожоге кожи жидким бромом рекомендуется промыть пострадавшее место разбавленным раствором аммиака.
Соединения и о д а играют важную роль в регулировании обмена веществ. У животных организмов иод накапливается главным образом в щитовидной железе (аналогично ведет себя и вводимый в организм астат). Тело человека содержит около 25 мг иода, из которых примерно 15 мг находится в щитовидной железе. Из обычных продуктов питания наиболее богаты иодом лук и морская рыба. Недостаток иода служит причиной болезни, известной под названием «зоба». Болезнью этой иногда страдает поголовно все население тех местностей (главным образом удаленных от моря возвышенностей), в которых воздух, вода и пища содержат слишком мало иода. Ежедневное потребление небольших — порядка 0,1 мг — доз иодидов (в виде примеси к поваренной соли) позволяет полностью избавиться от этой болезни. В Китае больных зобом издавна лечили золой морских губок (которая содержит до 8,5% иода). При добавлении в пищу иодсодержащих водорослей у коров увеличивается удой молока, а у овец быстрее растет шерсть. Отмечено также благотворное влияние небольших доз иодистых соединений на яйценоскость кур, откорм свиней и т. д.
Широко применяемая «иодная настойка» может быть приготовлена смешиванием в равных долях 10 %-ного раствора иода в спирте (95 %) и 4 %-ного водного раствора KI. Добавка иодистого калия повышает устойчивость жидкости при хранении. Следует отметить, что не только сам иод, но и многие его соединения (в частности, KI) хорошо всасываются организмом даже через неповрежденную кожу. Прием иодной настойки внутрь (1-5 капель на молоке) назначается иногда при атеросклерозе. Избыточное поступление иода в организм может вызвать некоторые неприятные явления (насморк, кожные сыпи и т. д.), исчезающие при прекращении приема иода.
Растворимость брома в воде составляет около 35 г, а иода — 0,3 г на литр. Оба эти галогена (и астат) гораздо лучше растворяются в различных органических растворителях.
Растворимость иода в воде сильно возрастает с повышением температуры и при 100 °С достигает 3,3 г/л. Органические жидкости растворяют его значительно лучше воды, как то видно из приводимых ниже примерных данных (в вес.% при обычных условиях):
С2H5OH | (C2H5)2O | C6H6 | CHCl3 | CCl4 | CS2 | ||
| 20 | 24 | 12 | 2,5 | 2,5 | 13 |
Растворы иода в разных растворителях имеют различные окраски: фиолетовую, красную, коричневую и промежуточных оттенков. Так как состоящие из свободных молекул I2 пары иода характеризуются сами по себе синей, а в смеси с воздухом фиолетовой окраской, наличие последней в растворе (например, в ССl4 или НГ) указывает на отсутствие заметной сольватации растворенных молекул иода. Напротив, коричневый цвет раствора (например, водного или спиртового) указывает на сильную сольватацию. В отличие от иода, цвет растворов брома почти не зависит от природы растворителя.
Благодаря лучшей, чем в воде, растворимости галоидов в органических растворителях, при соприкосновении водного раствора с органическим растворителем бульшая часть галогена переходит в последний. При этом галоген р а с п р е д е л я е т с я
между органическим растворителем и водой в строго определенных отношениях. Если в качестве примера взять бром и сероуглерод (СS2), то о т н о ш е н и е концентрации брома в сероуглеродной фазе к концентрации его в водной при различных общих количествах растворенного брома остается постоянным и равным примерно 80.
В этом постоянстве о т н о ш е н и я к о н ц е н т р а ц и й (точнее, отношения активностей) распределение между двумя несмешивающимися растворителями вещества заключается так называемый закон распределения. Он верен, однако, лишь в том случае, если распределяемое вещество в обеих фазах имеет один и тот же состав (например из молекул) и не вступает в прямое химическое взаимодействие с растворителем. Найденное отношение концентраций (в данном примере 80) называется коэффициентом распределения. Величина его (при постоянной температуре) характерна для данной системы: растворитель А — распределяемое вещество — растворитель Б. Например, при замене сероуглерода на ССl4 коэффициент распределения брома становится равным примерно 30. Распределение имеет большое техническое значение, так как часто позволяет избирательно извлекать (экстрагировать) то или иное вещество из раствора смеси веществ.
По своей наиболее характерной химической функции бром и иод являются о д н о в а л е н т н ы м и н е м е т а л л а м и. Некоторые числовые характеристики обоих элементов сопоставлены ниже с аналогичными данными для хлора и фтора (Г — общее обозначение галогена):
Молекула Г2 | Ядерное расстояние пм | Энергия Диссоциации кДж/моль | Атом Г | Эффективный радиус, пм | Сродство к электрону, кДж/моль | Ион Г | Эффективный радиус, пм | Энергия гидратации, кДж/моль |
F2 | 142 | 159 | F | 71 | 339 | F- | 133 | 485 |
Cl2 | 198 | 242 | Cl | 99 | 355 | Cl- | 181 | 351 |
Br2 | 229 | 192 | Br | 114 | 330 | Br- | 196 | 318 |
I2 | 267 | 150 | I | 133 | 301 | I- | 220 | 280 |
Химическая активность брома и иода меньше, чем у хлора, но все же велика. Со многими металлами и некоторыми неметаллами (например, фосфором) они способны взаимодействовать в обычных условиях. При этом бром по активности мало уступает хлору, тогда как иод отличается от него уже значительно.
Подобно атомам фтора и хлора, в основном состоянии атомы брома (4s24р5) и иода (5s25р5) одновалентны.
При выводе количественных характеристик сравнительной металлоидной активности галоидов в отсутствие воды вместо энергий гидратации должны учитываться энергии связей (в ковалентных системах) или энергии кристаллических решеток (в ионных системах). Как показывает приводимое ниже примерное сопоставление, все эти величины изменяются приблизительно однотипно:
| F | Cl | Br | I | |
Энергии гидратации ионов Г-, кДж/моль | 485 | 351 | 318 | 280 |
Энергии связей С-Г, кДж/моль | 485 | 339 | 284 | 231 |
| Энергии решеток NaГ, кДж/моль | 915 | 777 | 740 | 690 |
Поэтому общий характер изменения металлоидной активности по ряду F-С1-Вr-I остается неизменным.
На образовании и последующем термическом разложении летучих иодидов основано и о д и д н о е р а ф и н и р о в а н и е некоторых металлов (Сr, V, Тi и др.) Проводится оно в замкнутой системе путем взаимодействия иода с технически чистым образцом при 100-500 °С под давлением порядка 10-4 мм рт. ст., причем пары образующегося иодида тут же термически разлагаются на поверхности нагретой до 1300-1500 °С проволоки. Иод вновь вступает в реакцию, а вокруг проволоки постепенно наращивается стержень обрабатываемого металла, свободного от нелетучих при условиях опыта примесей.
Синтез НВr из элементов протекает при 200-300 °С с измеримой скоростью по следующим уравнениям:
Вr2 + 192 кДж = 2 Вr (первоначальное возбуждение),
Вr + Н2 = НBr + Н,
затем Н+ Вr2 = НBr + Вr и т. д.
В отличие от синтеза НСl вторая реакция затруднена из-за ее эндотермичности (71 кДж/моль), а обратная ей реакция
Н + НВг = Н2 + Вr
протекает легко. Поэтому возникающие цепи часто обрываются и процесс не приобретает взрывного характера. Так как реакция I + Н2 = НI + Н
еще более эндотермична (138 кДж/моль), синтез HI вообще не является цепной реакцией, а протекает по обычному бимолекулярному типу.
Взаимодействие брома с водородом происходит лишь при нагревании. Иод с водородом реагирует только при достаточно сильном нагревании и не полностью, так как начитает идти обратная реакция — разложение иодистого водорода. Оба галогеноводорода удобно получать разложением водой соответствующих галогенидных соединений фосфора по схеме:
РГ3 + 3 Н2О = Н3РО3 + 3 НГ
Реакция легко идет уже при обычной температуре.
Подобно хлористому водороду, HBr и HI представляют собой бесцветные газы, очень хорошо растворимые в воде. Некоторые их свойства сопоставлены со свойствами HF и HCl в приводимой ниже таблице и на рис. VII-12, на котором показаны также и радиусы ионов Г-. Как видно из рисунка, по ряду НI-НВr-НСl свойства изменяются весьма закономерно, тогда как при дальнейшем переходе к НF наблюдается более или менее резкий их скачок, иногда даже в направлении, обратном общему ходу. Обусловлено это сильной ассоциацией фтористого водорода, отсутствующей у его аналогов.
Энергии связей Н-Вr и Н-I равны соответственно 364 и 297 кДж/моль. Жидкие галоленоводороды характеризуются при температурах кипения плотностями 2,2 (НВr) и 2,8 (НI) г/см3 и теплотами испарения 17,6 и 19,6 кДж/моль. Как растворители, они похожи на НСl. Энергии диссоциации молекул НГ на свободные газообразные ионы Н+ и Г- составляют 1517(НF), 1359(НСl), 1317(НВr) и 1283(НI) кДж/моль. Теплота образования АtН из элементов оценивается в — 105 кДж/моль.
Судя по характеру изменения теплот образования гидрогалогенидов, их термическая устойчивость должна сильно уменьшаться от фтора к иоду. Действительно, распад НF на элементы становится заметен лишь выше 3500 °С, тогда как для других галоидоводородов имеем при 1000 °С следующие степени диссоциации: 0,0014(НС1), 0,5(НВг) и 33 % (НI). В органических растворителях (бензоле и т. п.) все гидрогалиды растворимы гораздо хуже, чем в воде.
Как и хлористый водород, НВr и НI образуют с водой азеотропные смеси, содержащие соответственно 47 % НВr (т. кип. 126 °С) и 57 % НI (т. кип. 127 °С). Для обоих галоидоводородов известны кристаллогидраты с 2, 3 и 4 молекулами воды. И для брома, и для иода были получены аналогичные соответствующему хлориду нестойкие производные типа [ХR4]НГ2, где R — органический радикал.
Увеличение электролитической диссоциации при переходе от НF к НI обусловлено, вероятно, уменьшением поверхностной плотности отрицательного заряда галоидов в связи с ростом их ионных радиусов.
В н е в о д н ы х растворителях галоидоводороды большей частью ведут себя как неэлектролиты или слабые электролиты. При этом обычно наблюдается гораздо более резкое усиление ионизации по мере повышения атомного номера галоида, чем в водных растворах. Так, в пиридине константы диссоциации галогеноводородов имеют следующие значения: 3·10-9 — (НF), 4·10-6 (НСl), 1·10-4 (HBr), 3·10-3 — (НI).
| Галогеноводород | Теплота образования из элементов, кДж/моль | Ядерное расстояние, пм | Длина молекулярного диполя, пм | Тпл, °С | Ткип, °С | Растворимость в воде моль/л при 10 °С | Степень диссоциации в 0,1 н. растворе, % |
| HF | 272 | 92 | 36 | -83 | +19,5 | Ґ | 9,0 |
| HCl | 92 | 128 | 23 | -114 | -85 | 14 | 92.6 |
| HBr | 33 | 141 | 17 | -87 | -67 | 15 | 93,5 |
| HI | -25 | 162 | 9 | -51 | -35 | 12 | 95,0 |
По химическим свойствам НВr и НI очень похожи на хлористый водород. Подобно последнему в безводном состоянии они не действуют на большинство металлов, а в водных растворах дают очень сильные бромистоводородную и иодистоводородную кислоты. Соли первой носят название б р о м и с т ы х или б р о м и д о в, второй — и о д и с т ы х или и о д и д о в (а производные галоидоводородных кислот вообще — г а л о г е н и д о в или га л и д о в). Растворимость бромидов и иодидов в большинстве случаев подобна растворимости соответствующих хлоридов. Возможность существования в виде отрицательно одновалентного иона установлена и для астата.
Существенное различие между НI, НВr и НСl наблюдается в их отношении к окислителям. Молекулярный кислород постепенно окисляет иодистоводородную кислоту уже при обычной температуре (причем под действием света реакция сильно ускоряется):
О2 + 4 НI = 2 Н2О + I2
Бромистоводородная кислота взаимодействует с ним гораздо медленнее, а соляная вовсе не окисляется молекулярным кислородом. Так как, однако, соляная кислота способна окисляться под действием MnО2 и т. п., из изложенного следует, что галоидоводороды (кроме НF) могут служить в качестве веществ, отнимающих кислород, т. е. в качестве в о с с т а н о в и т е л е й, причем наиболее активным в этом отношении является НI. Газообразный иодистый водород способен даже гореть в кислороде (с образованием Н2О и I2). Легкая окисляемость в растворах характерна и для производных отрицательно одновалентного астата.
Получение растворов иодистоводородной кислоты (вплоть до 50 %-ной концентрации) удобно вести, пропуская Н2S в водную суспензию иода. Реакция идет по схеме:
I2 + Н2S = 2 НI + S
Для предохранения водных растворов от окисления кислородом воздуха рекомендуется добавлять к ним небольшое количество красного фосфора (1 г/л), который, будучи практически нерастворимым в иодистоводородной кислоте, вместе с тем тотчас переводит образующийся при окислении свободный иод снова в НI.
Выделяющийся при частичном окислении иодистоводородной кислоты свободный иод не осаждается, а остается в растворе вследствие взаимодействия с избытком ионов I’ по схеме: I’ + I2 = I3’ + 16,7 кДж/моль. Аналогично могут возникнуть ионы Вr3’ и СI3’, а также ионы Г3’ образованные разными галоидами (кроме фтора). Образующийся в растворе ион Г3’ находится при этом в равновесии с продуктами своего распада: Г3’ Ы Г’ + Г2. Устойчивость ионов Г3’, зависит от природы галоида и характеризуется следующими значениями констант равновесия:
[Г3’]/[Г2]·[Г’] = K Г Сl Br I
K 0,2 16 700
Как видно из приведенных данных, по ряду С1-Вг-I устойчивость ионов Г3’ быстро возрастает. Разбавление растворов и нагревание благоприятствуют смещению равновесий вправо, большая концентрация Г’ — влево. Результатом существования подобных равновесий является более высокая растворимость свободных галоидов в растворах галогенидов по сравнению с чистой водой.
Система 3 I’ Ы I3’ + 2 е- часто служит рабочей средой х е м о т р о н о в — электрохимических установок для разностороннего оперирования со слабыми электрическими токами. Показанный на рис. VII-18 простейший хемотрон представляет собой небольшой замкнутый сосуд, заполненный раствором КI с незначительной добавкой свободного иода (т. е. содержит много ионов I’ и мало ионов I3-). Из двух впаянных платиновых электродов линейный (А) имеет малую рабочую поверхность, а сетчатый (Б) — большую. При включении тока в такой установке идут реакции:
3 I’ - 2 е- = I3’ — у анода и 2 е-+ I3’ = 3 I’ — у катода.
Если анодом является электрод А, а катодом — Б, то ионов I’ около первого много (благодаря их высокой концентрации в растворе), ионов I3’ около второго электрода тоже много (благодаря его большой поверхности), и ток свободно идет. Напротив, имеющийся около к а т о д а А небольшой запас ионов I3’, почти мгновенно исчерпывается, и ток практически прерывается. Рассматриваемая установка может, следовательно, служить выпрямителем слабых переменных токов низких частот, вообще же различные варианты хемотронов находят самое разнообразное техническое использование (например, в системах управления ракетными двигателями).
41) В зависимости от природы галоида, константы равновесия гидролиза имеют следующие значения:
[Н•]·[Г’]·[НОГ]/[Г2] = K 3·10-4 4·10-9 5·10-13
Г Cl Br I
В щелочной среде действительна иная трактовка гидролиза свободных галоидов, а именно по схеме:
Г2 + ОН’ Ы НОГ + Г’
При рассмотрении к и с л о р о д н ы х соединений брома и иода, как и в случае хлора, удобно исходить из обратимой реакции
Г2 + Н2О Ы НГ + НОГ
равновесие которой при переходе от хлора к брому и затем иоду все более смещается влево.
Растворы бромистоватистой (HOBr) и иодноватистой (HOI) кислот могут быть получены аналогично хлорноватистой кислоте. Обе кислоты являются н е у с т о й ч и в ы м и соединениями и с и л ь н ы м и о к и с л и т е л я м и. По ряду HOCl-HOBr-HOI и устойчивость и окислительная активность уменьшается.
В том же направлении от хлора к иоду, ослабляется и кислотный характер соединений НОГ. Бромноватистая кислота является уже очень слабой, тогда как иодноватистая обладает амфотерными свойствами. Обе кислоты известны только в разбавленных растворах желтоватой или зеленоватой окраски со своеобразными запахами.
42) Вероятно, удобным путем получения бромноватистой кислоты могла бы быть реакция по схеме:
Ag2SO4 + Вr2 + Ва(ОН)2 = 2 АgВrЇ + ВаSO4Ї + 2 НОВг
Перегонку растворов НОВr (К = 2·10-9) можно производить только под уменьшенным давлением (ниже +30 °С), а НОI без разложения вообще не перегоняется. Обе кислоты известны лишь в растворах (НОВr — до 30 %-ной концентрации). Особенно неустойчивая иодноватистая кислота может быть несколько стабилизирована добавлением иода ( в результате равновесия НOI + I’ Ы НOI2). Константа диссоциации НOI по кислотному типу (К = 2·10-11) даже меньше, чем по основному (3·10-10). Для реакции по уравнению
Н2О + Н2OI-Ы Н3О· + НOI
было получено значение константы равновесия К = 3·10-2. Это значит, что при [Н3О·] = 1 (и отсутствии ионов I’) более трети всего растворенного количества НOI находится в форме ионов Н2OI• (т. е. I•). С возможностью аналогичной основной диссоциации приходится считаться и у НОВr, и даже у НОСl.
Из солей обеих кислот в твердом состоянии были выделены только KOВr·3Н2О и кристаллогидраты NаОВr с 5 и 7 молекулами воды. Все эти светло-желтые соли очень неустойчивы, а при нагревании (или подкислении раствора) тотчас распадаются на соответствующие бромид и бромат.
43) Термическим разложением LiВгО3 при 200 °С был получен бромит лития — LiВrО3. Он представляет собой белый порошок, уже в присутствии следов воды разлагающийся по уравнению
3 LiВrО2 = LiВr + 2 LiВrО3
а при температуре плавления (225 °С) распадающийся на LiВr и O2. Аналогичные свойства характерны и для получаемого подобным же образом Ва(ВrО2)2.
44) При низких температурах (порядка -50 °С) бром окисляется озоном но реакции:
4 О3 + 3 Вr2 = 6 ВrО2
Образующийся диоксид брома (теплота образования из элементов — 54 кДж/моль) представляет собой светло-желтое твердое вещество, устойчивое лишь ниже -40 °С. Одним из продуктов ее термического разложения в вакууме является коричневый гемиоксид брома (Вr2О), плавящийся при -17 °С (с разложением) и дающий с водой НОВr. Гемиоксид брома частично образуется также при действии брома на сухой оксид ртути или его взвесь в СС14. Он устойчив лишь ниже -40 °С. Аналогичный оксид иода известен только в форме оранжево-красного двойного соединения с пиридином — I2O·4С5Н5N.
Помимо окислительного распада, для HOBr и HOI очень характерны реакции по схеме:
3 НОГ = 2 НГ + НГО3
ведущие к образованию бромноватой (HBrO3) или иодноватой (HIO3) кислоты. Первая известна только в растворах, а вторая может быть выделена в виде легкорастворимых кристаллов. Обе кислоты бесцветны.
Бромноватая кислота очень похожа по свойствам на HClO3, тогда как и окислительные, и кислотные свойства иодноватой выражены значительно слабее. По рядуHClO3-HBrO3-HIO3 растворимость солей, как правило, уменьшается. Подобно хлоратам, броматы и иодаты в щелочных и нейтральных средах окислителями не являются.
45) Скорость реакции 3 НОГ = 2 НГ + НГО3 при переходе от хлора к брому и затем иоду быстро возрастает. Для брома было экспериментально установлено, что она максимальна при равной концентрации ОВr’ и НОВr. Это позволяет предполагать активное участие в процессе молекул изобромноватистой кислоты — НВгО. И у брома, и у иода реакции протекают, вероятно, через промежуточное образование ионов ГО2’, однако аналогичные хлористой кислоте и хлоритам производные обоих элементов неизвестны. На приведенный выше основной процесс сильно налагается взаимодействие между НГ и НОГ. Поэтому общее уравнение разложения бромноватистой и иодноватистой кислот приближенно имеет вид:
5 НОГ = НГО3 + 2 Г2 + 2 Н2О
46) Растворы бромноватой кислоты могут быть получены, в частности, по реакции:
5 АgВrО3 + 3 Вr2 + 3 Н2О = 5 АgВr + 6 НВгО3
Концентрировать их удается лишь до 50 %-ного содержания (т. е. приблизительно до состава НВrО3·7H2O). И окислительные, и кислотные свойства НВrО3 приблизительно таковы же, как у НСlO3. Для иона ВrО3- даются значения d(ВrО) = 178 пм и РОВгО = 112°.
47) Иодноватая кислота образуется, в частности, под действие хлора, на водную суспензию иода по реакции
I2 + 5 Сl2 + 6 Н2О = 2 Н2O + 10 HCl
Поэтому при добавлении к раствору иодистой соли избытка хлорной воды появляющаяся вначале окраска иода затем вновь исчезает.
Для получения НIO3 (К = 0,2) обычно пользуются взаимодействием иода с крепкой азотной кислотой:
I2 + 10 НNО3 = 2 НIO3 + 10 NО2 + 4 H2O
Выделяющиеся окcиды азота удаляют пропусканием сквозь жидкость струи воздуха. Из сконцентрированного раствора при охлаждении осаждаются бесцветные кристаллы НIO3, плавящиеся при 110 °С (с переходом в НIO3·I2О5) и расплывающиеся на воздухе. Для молекулы НIO3 даются значения d(IO) = 180 пм (две связи) и 190 пм (одна связь), РOIO = 98°, а для иона IO3-, значения d(IO) = 182 пм и РOIO = 97°. В растворах иодноватой кислоты имеет место равновесие nНIO3 = (НIO3)3, где n = 2 или 3.
48) Растворимость производящихся от кислот НГО3 солей по ряду Сl-Br-I обычно уменьшается. Примером могут служить приводимые ниже данные (моль на литр Н2О при 20 °С):
NaClO3 | NaBrO3 | NaIO3 | KClO3 | KBrO3 | KIO3 | ||
| 9,2 | 2,3 | 0,46 | 0,58 | 0,41 | 0,38 |
В противоположность НСlO3 и НВrО3 для иодноватой кислоты, характерна совместная кристаллизация с ее солями. Известны NаIO3·2HIO3, КIO3·НIO3, KIO3·2НIO3 и т. д. Получены были также некоторые продукты присоединения к иодатам иодноватого ангидрида, например КIO3·I2О5 (т. пл. 316 °С).
Подобные соли иногда рассматривают как производные «трииодноватой» кислоты — НI3O8. Доводом в пользу такой трактовки может служить возможность получения свободной НI3O8 как путем частичного термического разложения НIO3, так и путем ее перекристаллизации из концентрированной НNО3. Однако «молекула» НI3O8 слагается из о т д е л ь н ы х молекул НIO3 и I2O5, между иодными и кислородными атомами которых существует лишь сильное м е ж м о л е к у л я р н о е взаимодействие.
Осторожным обезвоживанием HIO3 может быть получен белый порошок иодноватого ангидрида — I2O5. Он обладает сильными окислительными свойствами, а с водой вновь дает иодную кислоту.
49-52
Соли бромной кислоты (HBrO4) образуются при окислении броматов фтором в щелочной среде:
NaBrO3 + F2 + 2 NaOH = 2 NaF + NaBrO4 + H2O
Сама кислота по силе близка к хлорной, но гораздо менее устойчива (известна только в растворе) и является более сильным окислителем. Ее соли (перброматы) похожи по свойствам на перхлораты.
Иодная кислота (HIO4) может быть получена электролизом раствораHIO3 [по схеме H2O + HIO3 = H2(катод) + HIO4(анод)]. Выделяется она в виде бесцветного кристаллогидрата HIO4·2H2O. Кислотные свойства HIO4 выражены несравненно слабее, чем у HClO4, а окислительные, наоборот, гораздо более отчетливо. Большинство солей иодной кислоты (п е р и о д а т о в) малорастворимо в воде.
53-60
Как видно из рассмотренного выше материала, аналогия брома и иода с хлором в их кислородных соединениях выражена уже далеко не столь полно, как в водородных: закономерный характер изменения свойств при переходе по ряду Cl-Br-I здесь ограничивается главным образом кислотами типов НОГ и НГО3 и их солями. О кислородных соединениях астата известно лишь, что они существуют, причем высшая степень окисления отвечает иону AtO3-, т. е. степени окисления +5.
61
ДОПОЛНЕНИЯ
49) В отличие от оксидов других галоидов, I2O5 является экзотермичным соединением (теплота образования 184 кДж/моль). Практически он может быть получен постепенным нагреванием НIO3 до 120 °С с последующим длительным выдерживанием при этой температуре. Кристаллы иодноватого ангидрида слагаются из молекул O2I-O-IO2 со значениями d(OI) = 177ё183 пм, РOIO = 93ё102° для концевых частей и d(IO) = 192ё195 пм, РIOI = 139° — для центральной части. Продажный препарат обычно имеет розоватый или желтоватый оттенок (обусловленный следами свободного иода). Продажный ангидрид постепенно разлагается на свету и очень гигроскопичен. Применяется он главным образом при газовом анализе для определения монооксида углерода (основанного на реакции I2O5 + 5 СО = 5 СО2 + I2).
50) При действии тлеющего разряда на смесь паров брома с избытком охлажденного кислорода образуется триоксид брома — ВrО3 (вероятно, в димерной форме — Вr2О6). Оксид этот (которому ранее приписывали формулу Вr2О6) представляет собой бесцветное кристаллическое вещество, устойчивое лишь ниже -70 °С. С водой он образует, по-видимому, две кислоты — НBrО3 и НВrO4, из которых последняя тотчас же разлагается на HBrО3 и кислород.
Вместе с тем взаимодействием Вr2 с избытком озона были получены Br3O8 и Вr2О5, но получить таким путем Вr2О6 не удалось. Вопрос о высших оксидах брома остается, таким образом неясным.
53) Несмотря на неоднократные попытки, бромную кислоту впервые удалось получить только в 1968 г. При обычных условиях ее бесцветный раствор устойчив приблизительно до 6 М концентрации (55 %-ного содержания). Более крепкие растворы при хранении желтеют (вследствие восстановления НВrO4 до свободного брома)/ Как окислитель бромная кислота значительно сильнее хлорной, но окисляет она медленно (как и хлорная). Растворимость КВrО4, при комнатной температуре составляет около 0,2 М, т. е. несколько больше, чем у КСlO4. Ион ВrО4-, представляет собой тетраэдр с d(ВrО) = 161 пм. Пербромат калия термически устойчив до 280 °С (против 610 °С для КСlO4). Получен и пербромат аммония — NН4ВrO4.
54) Как кислота НIO4 (K = 3·10-2) слабее иодноватой. Наоборот, как окислитель она более активна, чем HIO3 (но менее, чем НOI). Весьма интересно отношение НIO4 к воде. При их взаимодействии в зависимости от условий может образоваться несколько соединений общей формулы (НIO4)n·(Н2О)m. Во всех таких соединениях водороды воды способны з а м е щ а т ь с я на м е т а л л также, как и водород самой НIO4. В связи с этим соединения подобного типа обычно рассматривают как с л о ж н ы е к и с л о т ы и приписывают им следующие формулы: НIO4 (n=1, m=0), Н3IO5 (n=1, m=1), Н4I2O9 (n=2, m=1), Н5IO6 (n=1, m=2). Например, были получены К4I2O9 и следующие серебряные соли: оранжевая АgIO4, красная АgНIO5, черная Аg3IO5, зеленовато-желтая Аg2Н3IO6 и черная Аg5IO6.
55) При взаимодействии НIO4 с 65 %-ным олеумом образуется оранжевое твердое вещество. Судя по результатам анализа, оно представляет собой иодный ангидрид — I2O7. Свойства его пока не изучены. Двойным соединением I2O7·I2O5 является, вероятно, желтый продукт термического разложения Н5IO6 в вакууме при 110 °С.
56) Соли иодных кислот, как правило, труднорастворимы в воде. Некоторые из них весьма термически устойчивы (например, Nа5IO6 выдерживает без разложения нагревание до 800 °С). Получают периодаты обычно действием хлора в щелочной среде на соли иодноватой кислоты (например, по реакции
NaIO3 + 4 NаОН + Сl2 = Nа3Н2IO6 + 2 NаСl + Н2О
или же электролизом растворов солей HIO3.
57) Сообщалось, что термический распад Nа2Н3IO6 около 200 °С ведет к образованию Nа2IO4. Магнитные свойства препарата подтверждают как будто, что это вещество является производным шестивалентного иода. Оно устойчиво до 370 °С, а водой тотчас разлагается на иодат и периодат. Подобным же образом были получены некоторые другие соли, предположительно также являющиеся производными шестивалентного иода.
60) Кроме рассмотренных выше кислородных соединений брома и иода, известны еще некоторые. Из них наиболее интересны производные т р е х в а л е н т н о г о иода, в которых он играет роль металла. Например, были получены устойчивый лишь ниже 0 °С желтый I(NО3)3, желтый IРO4, желто-зеленый I(ClO4)3·2Н2О и бесцветный I(СН3СОО)3. При электролизе последней соли иод выделяется на катоде, чем и доказывается его положительный заряд. Из аналогичных производных брома известен бесцветный Br(NO3)3.
Солеобразные производные о д н о в а л е н т н ы х иода и брома очень неустойчивы сами по себе, но некоторые из них довольно устойчивы в виде двойных соединений с пиридином. Например, желтый INО3 разлагается уже выше -5 °С, тогда как бесцветный INО3·2С5H5N плавится при 138 °С без разложения. Сходные свойства имеют желтый ВrNО3 (т. пл. -42 °С) и бесцветный ВrNО3·2С5Н5N (т. пл. 80 °С). Известны также аналогичные нитратам по составу перхлораты и производящиеся от одновалентного иода соли некоторых органических кислот. Наиболее интересным из этих производных Вr+ является б р о м п е р х л о р а т, который был получен при -45 °С по реакции
Вr2 + 2 СlClO4 = Сl2 + 2 ВrСlO4
и представляет собой красную жидкость, еще не замерзающую при -78 °С и медленно разлагающуюся уже при -20 °С. Озонированием ВrNО3 был получен очень неустойчивый оранжевый ВrО2NО3.
Растворение смеси 2 I2 + 3 I2O5 (что эквивалентно 5 I2O3) в концентрированной Н2SO4 ведет к образованию желтых расплывающихся на воздухе кристаллов (IO)2SO4. При обработке дымящей Н2SО4 они белеют, по-видимому, вследствие перехода в (IO)НSО4. Обработка сульфатов иода водой сопровождается выделением I2 и желтого труднорастворимого порошка состава I2O4. Оксид этот, разлагающийся выше 100 °С на I2 и I2O5, следует рассматривать как о с н о в н у ю иодноватокислую соль трехвалентного иода — (IO)IO3.
При обработке иода озоном образуется желтоватый расплывающийся на воздухе (с разложением) порошок состава I4O9. По-видимому, он представляет собой среднюю иодноватокислую соль трехвалентного иода — I(IO3)3. Выше 75 °С иодтрииодит разлагается с выделением иода.
61) Астат несколько менее летуч, чем иод, и из разбавленных азотнокислых растворов не отгоняется (в отличие от иода). Сероводородом в солянокислой среде он осаждается вместе с Вi2S3 и Sb2S3, а обработка осадка сернистым аммонием частично переводит At в раствор. Состав образующихся при этом его соединений не установлен. Самый концентрированный из подвергавшихся исследованиям раствор соединений астата был относительно него 10-8 М.
Наиболее сильными окислителями (в частности, НОС1) астат окисляется до иона АtО3’. Известна и другая, более низкая положительная валентность Аt, возникающая при его обработке менее сильными окислителями (Вr2, НNО3 и т. д.). По-видимому, в этих условиях образуется ион ОАt’. Установлена также «возможность замещения астатом (А1') иода в его производных типа IХ·2С5Н5N (где Х = NО3 или СlO4). Раствор FеSO4 восстанавливает окисленные состояния Аt до элементарного. Действием Zn в кислой среде (или SnСl2 в щелочной) астат может быть далее восстановлен до иона Аt’. Последний легко вновь окисляется до элементарного Аt.
ПЯТАЯ ГРУППА ПЕРИОДИЧЕСКОЙ СИСТЕМЫ
По электронным структурам нейтральных атомов рассматриваемая группа может быть разделена на две подгруппы. Одна из них включает азот, фосфор, мышьяк и его аналоги, вторая — ванадий и его аналоги.
Так как атомы N-Bi имеют во внешнем слое по пять электронов, можно ожидать у них тенденцию к дополнению этого слоя до октета. Однако эта тенденция должна проявляться менее резко, чем у соответствующих элементов седьмой (F-At) и шестой (О-Ро) групп, которым до восьмиэлектронной конфигурации не хватает соответственно лишь по одному или по два электрона. В связи с этим неметаллические свойства, например, у фосфора выражены слабее, чем у хлора и серы. С другой стороны, отдача электронов нейтральными атомами должна происходить в V группе легче, а устойчивость кислородных соединений должна быть больше, чем у соответствующих элементов VII и VI групп.
Подобно элементам подгруппы хрома, ванадий и его аналоги характеризуются наличием во внешнем слое не более двух электронов, что обуславливает отсутствие тенденции к их дальнейшему присоединению. Вместе с тем можно ожидать, что в производных высшей валентности ванадий и его аналоги будут иметь значительное сходство с фосфором.
Азот
Общее содержание азота в земной коре оценивается в 0,03%. Наибольшая его часть (около 4·1015 т) сосредоточена в атмосфере, основную массу которой (75,6 вес. %) и составляет свободный азот (N2). Сложные органические производные азота входят в состав всех живых организмов. В результате отмирания этих организмов и тления их останков образуются более простые азотные соединения, которые при благоприятных условиях (главным образом при отсутствии влаги) могут накапливаться. Именно такого происхождения природные залежи NaNO3 в Чили, имеющие промышленное значение как один из источников получения связанного азота.
Первые указания на азот как особое вещество были получены в 1772 г. Природный азот состоит из смеси двух изотопов — 14N (99,63%) и 15N (0,37%).
Атом азота в основном состоянии имеет структуру внешнего электронного слоя 2s22p3 и трёхвалентен.
Так как свободный азот содержится в атмосфере, получение его сводится к отделению от кислорода и других составных частей воздуха. Технически оно осуществляется постепенным испарением (“фракционной перегонкой”) жидкого воздуха в специальных установках. При этом одновременно получаются кислород и инертные газы. В лабораторных условиях азот может быть получен по реакции:
NH4NO2 = 2 H2O + N2 + 334 кДж,
которая легко протекает при нагревании концентрированного раствора нитрита аммония.
Так как разложение NH4NO2 сильно экзотермично, нагревание (примерно до 70 °С) необходимо лишь для начала реакции. В дальнейшем приходится, наоборот, охлаждать реакционный сосуд, чтобы избежать слишком бурного протекания процесса. Вместо нитрита аммония можно пользоваться смесью NaNO2 и NH4Cl, так как при взаимодействии между ними по реакции:
NaNO2 + NH4Cl Ы NaCl + NH4NO2
частично образуется NH4NO2, последующее разложение которого (лучше идущее в слабокислой среде) всё время смещает равновесие вправо. Практически удобнее медленно пускать по каплям насыщенный раствор NaNO2 в нагретый насыщенный раствор (NH4)2SO4. Выделяющийся газ освобождают от следов NH3, NO и О2 последовательным пропусканием сквозь растворы H2SO4 и FeSO4, а затем над накалённой медью, после чего азот подвергают сушке.
Азот может быть получен также нагреванием смеси грубо измельчённых K2Cr2O7 (2 вес. ч.) и (NH4)2SO4 (1 вес. ч.). Смеcь эта разлагается аналогично бихромату аммония, но реакция идёт лишь при нагревании. Наиболее чистый азот получают термическим разложением при 300 °С тщательно высушенного азида натрия по схеме:
2 NaN3 = 2 Na + 3 N2.
Содержащие азот баллоны должны иметь чёрную окраску с жёлтой надписью “Азот”, подчёркнутой коричневой полосой.
При обычных условиях азот представляет собой бесцветный не имеющий запаха газ. Бесцветен от и в жидком, и в твёрдом состоянии. Точка плавления азота лежит при –210 °С, точка кипения при –196 °С. Растворимость его в воде мала — около 2 объёмн.%. Молекула азота двухатомна и заметно не распадается на атомы даже при очень высоких температурах.
В молекуле N2 осуществляется тройная связь между атомами азота. Она характеризуется ядерным расстоянием d(NN) = 109,5 пм и энергией диссоциации 945 кДж/моль.
Экспериментально установлено, что заметная термическая диссоциация молекул N2 на атомы до 3000 °С не наступает. Под обычным давлением степень диссоциации не превышает нескольких процентов даже при 5000 °С. Фотохимическая диссоциация молекул N2 протекает лишь в высоких слоях атмосферы. Искусственное получение атомарного азота может быть осуществлено путём пропускания газообразного N2 (под сильно уменьшенным давлением) сквозь поле высокочастотного электрического разряда. Так как энергии активации реакций с участием свободных атомов обычно весьма малы (часто близки к нулю), атомный азот гораздо активнее молекулярного: уже при обычной температуре он непосредственно соединяется с S, P, As, также с Hg и рядом других металлов.
Если азот, содержащий смесь молекул и атомов, направлять на охлаждаемую жидким гелием поверхность, происходит его мгновенное замораживание. Оно сопровождается ярким зелёным свечением, которое переходит затем в синие вспышки. И то, и другое обусловлено выделением энергии при частично происходящем обратном соединении (рекомбинации) нормальных и возбуждённых атомов азота в молекулы. Однако многие атомы оказываются при замораживании отделёнными друг от друга молекулами N2. В таком “замороженном” состоянии они могут некоторое время (несколько часов) сохраняться. Если содержащее их твёрдое вещество нагреть, происходит рекомбинация атомов, сопровождающаяся вспышкой синего цвета.
Аналогично азоту может быть осуществлено “замораживание” атомов других газов, а также более сложных свободных радикалов (например, ОН). Исходную смесь частиц обычно разбавляют аргоном или каким-либо другим не взаимодействующим с данными активными частицами веществом, которое создаёт при замораживании скелетную сетку (матрицу), изолирующую такие частицы друг от друга. При получении свободных радикалов (особенно органических) чаще пользуются действием проникающих излучений на уже замороженное и сильно охлаждённое исходное вещество. Так как при рекомбинации некоторых свободных радикалов выделяется много энергии, проблема их получения и хранения представляет значительный интерес не только для химии (где они могут быть использованы как инициаторы реакций или участники процессов необычного типа), но и для реактивной техники.
Под обычным давлением твёрдый азот существует в двух аллотропических формах, точка перехода между которыми лежит при –238 °С (теплота перехода 0,25 кДж/моль). Теплота его плавления составляет лишь 0,84 кДж/моль, теплота испарения 5,43 кДж/моль. Критические температура и давление азота равны соответственно –147 °С и 33,5 атм.
Свободный азот химически весьма инертен. При обычных условиях он практически не реагирует с неметаллами и металлами (кроме Li). Нагревание увеличивает его химическую активность главным образом по отношению к металлам, с некоторыми из которых он соединяется, образуя нитриды (например, Mg3N2).
Свободный азот применяется главным образом для охлаждения и создания инертной атмосферы. Первое осуществляется жидким азотом, второе — газообразным. Было показано, что заполнение им (вместо воздуха) автомобильных шин ведёт к существенному повышению их срока службы.
Растворимость азота в воде под обычным давлением весьма мала, но с его повышением сильно возрастает, достигая при 1000 атм приблизительно 7 объёмов на один объём воды.
Наиболее характерным для азота валентным состояниям отвечают значения –3, 0, +3 и +5. Схема окислительно-восстановительных потенциалов (В), соответствующих переходам между ними:
—3 0 +3 +5
Кислая среда +0,27 +1,45 +0,94
Щелочная среда –0,74 +0,41 +0,01.
Жидкий азот потребляется преимущественно для глубокого охлаждения, газообразный — как исходный продукт для синтеза различных его производных. Соединения азота имеют громадное значение для биологии и разнообразных отраслей промышленности. Наибольшие их количества расходуются в качестве минеральных удобрений и при производстве взрывчатых веществ.
Аммиак. Перевод свободного азота воздуха в связанное состояние осуществляется главным образом путём синтеза аммиака:
N2 + 3 H2Ы 2 NH3 + 92 кДж.
Принцип смещения равновесия показывает, что наиболее выгодными для образования аммиака условиями являются возможно более низкая температура и возможно более высокое давление. Однако даже при 700 °С скорость реакции настолько мала (и следовательно, равновесие устанавливается так медленно), что не может быть и речи о её практическом использовании. Напротив, при более высоких температурах, когда равновесное состояние устанавливается быстро, ничтожно малым становится содержание аммиака в системе. Таким образом, техническое проведение рассматриваемого процесса оказывается как будто невозможным, так как, ускоряя достижение равновесия при помощи нагревания, мы одновременно смещаем его положение в невыгодную сторону.
Существует, однако, средство ускорить достижение равновесного состояния без одновременного смещения равновесия. Таким часто помогающим средством является подходящий катализатор. Подходящим катализатором является металлическое железо (с примесями Al2O3 и К2О). Процесс обычно ведут при температуре 400-600 °С (на катализаторе) и давлениях 100-1000 атм. После выделения аммиака из газовой смеси последняя вновь вводится в цикл.
В процессе поисков катализатора для синтеза аммиака было перепробовано около 20 тыс. различных веществ. Широко применяемый железный катализатор готовится обычно нагреванием тесной смеси FeO и Fe2O3 (содержащий небольшие примеси Fe, Al2O3 и КОН) в атмосфере состава 3Н2+N2. Так как Н2S, CO, CO2, водяной пар и кислород быстро “отравляют” катализатор, подаваемая к нему азотоводородная смесь должна быть тщательно освобождена от них. При правильном технологическом режиме катализатор бесперебойно работает в течение нескольких лет.
Для дальнейшего развития промышленности синтетического аммиака может оказаться существенным, что при давлениях в 2000 атм и выше синтез аммиака из азотоводородной смеси хорошо идёт и без специального катализатора. Практический выход аммиака при 850 °С и 4500 атм составляет 97%. Особенно важно то обстоятельство, что при сверхвысоких давлениях наличие в исходных газах различных примесей не влияет на ход процесса.
Синтез аммиака был практически реализован в 1913 г., когда таким путём удалось получить 7 т NH3. В настоящее время этот синтез является основным промышленным методом получения связанного азота с ежегодной мировой выработкой, исчисляемой десятками миллионов тонн.
Помимо прямого синтеза аммиака из элементов, некоторое промышленное значение для связывания азота воздуха имеет разработанный в 1905 г. цианамидный способ. Последний основан на том, что при 1000 °С карбид кальция (получаемый прокаливанием смеси извести и угля в электрической печи) реагирует со свободным азотом по уравнению:
СаС2 + N2 = CaCN2 + C + 293 кДж .
Полученный таким путём цианамид кальция (Са=N–CєN) представляет собой серый (от примеси углерода) порошок. При действии перегретого (т. е. нагретого выше 100 °С) водяного пара он разлагается с выделением аммиака:
СаСN2 + 3 H2O = CaCO3 + 2 NH3 + 222 кДж.
Разложение цианамида кальция водой медленно протекает при обычных температурах. Поэтому им можно пользоваться как азотным удобрением, внося его в почву задолго до посева. Наличие кальция делает его особенно пригодным для подзолистых почв. “Цианамид играет роль не только азотистого, но и известкового удобрения, причём известь является бесплатным приложением к азоту” (Д.Н. Прянишников).
В лабораторного условиях NH3 получают путём обработки твёрдого NH4Cl насыщенным раствором КОН. Выделившийся газ может осушен пропусканием сквозь сосуд с твёрдым КОН или со свежепрокаленным оксидом кальция (СаО). Применять для сушки H2SO4 и CaCl2 нельзя, так как аммиак образует с ними соединения.
Молекула NH3 имеет структуру треугольной пирамиды с атомом азота в вершине. Электроны связей Н-N довольно сильно смещены от водорода к азоту, поэтому молекула аммиака в целом характеризуется значительной полярностью.
Пирамидальная структура аммиака энергетически выгоднее плоской на 25 кДж/моль. Молекула полярна; связь N-H характеризуется энергией 389 кДж/моль, но для энергий последовательной диссоциации атомов водорода даются значения 435, 397 и 339 кДж/моль.
Под обычным давлением аммиак сжижается при –33 °С и затвердевает при –78 °С. Теплота плавления NH3 составляет 6 кДж/моль. Критическая температура аммиака 132 °С, критическое давление — 112 атм. Содержащие его баллоны должны быть окрашены в жёлтый цвет и иметь чёрную надпись “Аммиак”.
Аммиак представляет собой бесцветный газ с характерным резким запахом (“нашатырного спирта”). Растворимость его в воде больше, чем всех других газов: один объём воды поглощает при 0 °С около 1200, а при 20 °С — около 700 объёмов NH3. Продажный концентрированный раствор имеет обычно плотность 0,91 г/см3 и содержит 25 вес.% NH3 (т.е. близок к составу NH3·3H2O).
Интересным свойством молекул аммиака является их способность к структурной инверсии, т.е. к “выворачиванию наизнанку” путём прохождения атома азота сквозь образованную атомами водорода плоскость основания пирамиды. Потенциальный барьер этой инверсии равен 25 кДж/моль, осуществлять её могут лишь молекулы, достаточно богатые энергией. Скорость инверсии сравнительно невелика — она в 1000 раз меньше скорости ориентации молекул NH3 электрическим полем. Инверсия связана с излучением строго определённой частоты (n = 2,387·1010 с-1), на основе чего была создана аппаратура для очень точного измерения времени. Такие “молекулярные часы” позволили установить, что продолжительность земных суток ежегодно возрастает на 4,3·10-4 с.
Аммиак сильно раздражает слизистые оболочки уже при 0,5%-ном содержании его в воздухе. Острое отравление аммиаком вызывает поражения глаз и дыхательных путей, одышку и воспаление лёгких. Средствами первой помощи служат свежий воздух, обильное промывание глаз водой, вдыхание водяного пара. Хроническое отравление аммиаком вызывает расстройство пищеварения, катары верхних дыхательных путей и ослабление слуха. Предельно допустимой концентрацией NH3 в воздухе производственных помещений считается 0,02 мг/л. Смеси аммиака с воздухом, содержащие от 16 до 28 объёмн.% аммиака взрывоопасны.
При пропускании струи аммиака над нагретой CuO он окисляется до свободного азота. Окисление аммиака озоном ведёт к образованию NH4NO3. Интересно, что некоторое участие в таком окислении принимает, по-видимому, и смешанный с озоном обычный кислород.
Аммиак является хорошим горючим реактивного топлива. Подобно воде, жидкий аммиак сильно ассоциирован, главным образом за счёт образования Н-связей. Однако они сравнительно слабы (около 4,2 кДж/моль). Вязкость жидкого аммиака почти в семь раз меньше вязкости воды. Его плотность (0,68 и 0,61 г/см3 соответственно при –33 и +20 °С) также значительно меньше, чем у воды. Электрический ток жидкий аммиак практически не проводит, так как электролитическая диссоциация по схеме:
NH3 + NH3Ы NH4++ NH2-
ничтожно мала: ионное произведение [NH4+][NH2-] = 2·10-33 (при –50 °С).
С ассоциацией жидкого аммиака связана его большая теплота испарения (23,4 кДж/моль). Так как критическая температура аммиака лежит высоко (+132 °С) и при испарении его от окружающей среды отнимается много тепла, жидкий аммиак может служить рабочим веществом холодильных машин.
Выше 0 °С (под давлением) жидкий аммиак смешивается с водой в любых соотношениях. На крепких растворах воды в аммиаке при 30 °С было показано, что её ионизация мала. Так, для 9 М раствора имеем [NH4+][OH-]/[H2O] = 1·10-11.
Жидкий аммиак является хорошим растворителем для очень большого числа органических соединений, а также многих неорганических. Например, хорошо растворяется в жидком аммиаке элементарная сера, крепкие растворы которой имеют красный цвет [и ниже +18 °С содержат сольват S(NH3)2]. Из солей лучше других растворимы производные аммония и щелочных металлов, причём по ряду Cl-Br-I растворимость солей возрастает. Примерами могут служить следующие данные (г/100 г NH3 при 25 °С):
NH4Cl NH4Br NH4I KCl KВr KI AgCl AgBr AgI
103 238 369 0,04 13,5 182 0,83 5,9 207
Подобный же ход изменения растворимости галогенидов характерен и для ряда других катионов. Хорошо растворимы в жидком аммиаке также многие нитраты (и КМnO4). Напротив, оксиды, фториды, сульфаты и карбонаты, как правило, в нём нерастворимы.
Пользуясь различием растворимости солей в жидком NH3 и воде, можно иногда осуществлять обращение обычно наблюдаемых реакций ионного обмена. Например, равновесие по схеме:
2 AgNO3 + BaBr2Ы 2 AgBr + Ba(NO3)2
в водной среде практически нацело смещается вправо (из-за нерастворимости АgBr), а в аммиачной среде — влево (из-за нерастворимости ВаВr2).
Характерным свойством аммиака как ионизирующего растворителя является его резко выраженное выравнивающее влияние на диссоциацию различных электролитов. Например, несоизмеримые друг с другом по диссоциации в водной среде HClO4 и HCN в жидком аммиаке характеризуются почти одинаковыми константами диссоциации (5·10-3 и 2·10-3). Соли ведут себя в жидком аммиаке как электролиты средней силы или слабые (например, К = 2·10-3 для КВr). Хлориды обычно бывают диссоциированы несколько менее, а иодиды — несколько более, соответствующих бромидов.
Особенностью жидкого аммиака является его способность растворять наиболее активные металлы, причём последние подвергаются ионизации. Например, разбавленный раствор металлического натрия имеет синий цвет, проводит электрический ток подобно растворам электролитов и содержит катионы Na+ (cольватированные аммиаком) и анионы (NH3)x-. Центральной частью такого сложного аниона является свободный электрон, находящийся в поляризационном взаимодействии с окружающей средой (полярон). При более высоких концентрациях Na его раствор приобретает вид бронзы и проявляет металлическую электропроводность, т. е. наряду с сольватированным аммиаком содержатся и свободные электроны. Ниже –42 °С синяя и бронзовая фазы способны сосуществовать, не смешиваясь. Длительное хранение растворов натрия в жидком аммиаке сопровождается их обесцвечиванием в результате очень медленной реакции по схеме:
2 Na + 2 NH3 = 2 NaNH2 + H2.
C цезием (растворимость 25 молей на 1000 г NH3 при –50 °С) аналогичная реакция протекает за несколько минут.
Растворённый в аммиаке металл имеет тенденцию к отщеплению валентных электронов, что создаёт возможность проведения своеобразных реакций вытеснения. Например, пользуясь растворимостью в жидком аммиаке КСl и нерастворимостью СаСl2, можно осуществить выделение калия кальцием по схеме:
2 КСl + Ca ® CaCl2 + 2 K.
Имеется интересное указание на то, что пропитка жидким аммиаком сильно повышает пластичность древесины. Это позволяет сравнительно легко придавать ей те или иные заданные формы, которые после удаления аммиака сохраняются.
Растворение аммиака в воде сопровождается выделением тепла (около 33 кДж/моль). Влияние температуры на растворимость иллюстрируется приводимыми ниже данными, показывающими число весовых частей NH3, поглощаемое одной весовой частью воды (под давлением аммиака, равным атмосферному):
Температура °С –30 0 10 30 50 80 100
Растворимость 2,78 0,87 0,63 0,40 0,23 0,15 0,07
Максимальной электропроводностью обладает при обычных условиях приблизительно 3 н раствор аммиака. Растворимость его в органических растворителях значительно меньше, чем в воде.
Для химической характеристики аммиака основное значение имеют реакции трёх типов: присоединения, замещения водорода и окисления.
Наиболее характерные для аммиака реакции присоединения. В частности, при действии его на многие соли легко образуются кристаллические аммиакаты состава СаСl2·8NH3, CuSO4·4NH3 и т.п., по характеру образования и устойчивости похожие на кристаллогидраты.
При растворении аммиака в воде происходит частичное образование гидроксида аммония:
NH3 + H2O Ы NH4OH
В этом соединении радикал аммоний (NH4) играет роль одновалентного металла. Поэтому электролитическая диссоциация NH4OH протекает по основному типу:
NH4OH Ы NH4• + OH’
Объединяя оба эти уравнения, получаем общее представление о равновесиях, имеющих место в водном растворе аммиака:
NH3 + H2O Ы NH4OH Ы NH4•+ OH’
Из-за наличия этих равновесий водный раствор аммиака (часто называемый просто “аммиаком”) имеет резкий запах. Ввиду того что концентрация ионов ОН’ в растворе невелика, NH4OH рассматривается как слабое основание. Гидроксид аммония является одним из важнейших химических реактивов, разбавленные растворы которого (“нашатырный спирт”) применяются также в медицине и домашнем хозяйстве (при стирке белья и выводе пятен).
Гидроксид аммония при обычных условиях совершенно неустойчив, однако в водных растворах аммиака он, несомненно, существует. Простым физическим растворением NH3 в воде нельзя объяснить резкое снижение давления его пара над раствором. Это явление может обуславливаться двумя основными причинами. Первая — ионизация молекул NH3, вследствие незначительности не может играть решающей роли. Вторая причина, играющая решающую роль, —гидратация молекул NH3, осуществляющаяся путём образования Н-связей по двум типам: Н3N·HOH и Н2О·НNH2. Так как водород более положительно поляризован в воде, а протонное сродство у азота выше, чем у кислорода, взаимодействие приводит в основном к образованию молекул Н3NHOH или NH4OH, т. е. гидроксида аммония.
Среднее время жизни индивидуальной молекулы H3NHOH в растворе аммиака очень мало (порядка 2·10–12 с). Такие молекулы всё время и разрушаются, и вновь возникают (равновесие NH3 + H2O Ы H3NHOH характеризуется значением К = 0,2). В связи с их малой устойчивостью существование гидроксида аммония — NH4OH — нередко вообще отрицается (даже в качестве промежуточного продукта при электролитической диссоциации приведённой выше). Между тем от NH4OH производятся все соли аммония, и уже тем самым это основание имеет право на существование в химической практике независимо от характера его внутримолекулярных связей и устойчивости.
Анализ данных по распределению NH3 между водой и органическими жидкостями показывает, что в гидратированной форме находится более 90% всего растворённого в воде аммиака. Для паровой фазы над водно-аммиачным раствором установлено наличие равновесия по схеме:
2 NH3 + H2O Ы 2 NH3·H2O + 75 кДж,
характеризующегося значением К = 1·10-4 при 20 °С.
Учитывая наличие в растворе одновременно с NH4OH также молекул NH3 (как гидратированных по второму типу, так и “просто” растворённых), выражение для константы диссоциации гидроксида аммония можно было бы написать в следующей уточнённой форме:
[NH4•][OH’]/[NH4OH + NH3] = 2·10-5
Однако уточнять его подобным образом применительно к данному случаю нет особых оснований, так как в аналогичном положении находится ряд других электролитических систем (например, SO2 + H2O). В нормальном водном растворе аммиака рН = 11,6, в децинормальном 11,1 и в сантинормальном 10,6.
Нашатырный спирт поступающий в продащу, содержит обычно около 10% аммиака. Он находит и медицинское применение. В частности, вдыхание его паров или приём внутрь (3-10 капель на рюмку воды) используется для снятия состояния сильного опьянения. Смазывание кожи нашатырным спиртом ослабляет действие укусов насекомых. Очень разбавленным нашатырным спиртом удобно протирать окна и мыть окрашенные масляной краской полы, более крепким — удалять следы от мух, чистить серебряные или никелированные предметы.
При выводе пятен хорошие результаты дают во многих случаях следующие составы (по объёму): а) 4 части нашатырного спирта, 5 частей эфира и 7 частей винного спирта (денатурата); б) 5 частей нашатырного спирта, 2 части бензина и 10 частей винного спирта; в) 10 частей нашатырного спирта, 7 частей винного спирта, 3 части хлороформа и 80 частей бензина; г) 5 частей нашатырного спирта, 3 части ацетона и 20 частей спиртового раствора мыла.
Попавшую на одежду масляную краску рекомендуется оттирать кусочками ваты, смоченными сперва скипидаром, а затем нашатырным спиртом. Для удаления чернильного пятна обычно достаточно обработать его нашатырным спиртом и смыть водой.
Добавление кислот ведёт к смещению приведённых выше равновесий вправо (ввиду связывания ионов ОН’) и к образованию солейаммония, по уравнению:
NH4OH + HCl = H2O + NH4Cl
Cоли эти образуются также при непосредственном взаимодействии аммиака с кислотами, которое сопровождается выделением тепла. Например, для газообразных NH3 и HCl имеем:
NH3 + HCl = NH4Cl + 176 кДж.
Интересно, что при полном отсутствии воды реакция эта не идёт.
Как сам ион аммония (NH4+), так и большинство его солей бесцветны. Почти все они хорошо растворимы в воде и в растворах сильно диссоциированы. Соли аммония играют важную роль при многих производственных процессах и широко используются в лабораторной практике.
Хлористыйаммоний (“нашатырь”) при высоких температурах реагирует с оксидами металлов, обнажая чистую металлическую поверхность. На этом основано его использование при пайке металлов. Он часто вводится в состав дымовых смесей (такая смесь может содержать, например 50 вес.% NH4Cl, 20 нафталина, 10 древесного угля и 20% КСlO3). В электротехнике NH4Cl употребляется для изготовления “сухих” гальванических элементов, в медицине он иногда прописывается для приёма внутрь (как отхаркивающее).
Взаимодействие NH4Cl с оксидами металлов при нагревании может протекать по двум схемам:
4 МеО + 2 NH4Cl = 4 H2O + N2 + MeCl2 + 3 Me и
MeO + 2 NH4Cl = MeCl2 + 2 NH3 + H2O.
В первом случае основным процессом является восстановление оксида аммиаком. Такое направление реакции характерно для оксидов сравнительно малоактивных металлов (например Сu). Во втором случае простая реакция обмена сопровождается возникновением летучего при нагревании хлорида.
Рассмотрим устройство одного из типов “сухого” гальванического элемента: катодом которому служит внешняя цинковая оболочка элемента, анодом — угольный стержень, вокруг которого находится смесь мелкораздробленных графита и MnO2, заключённая в оболочку из ткани. Промежуток между катодом и анодом заполнен влажной пастой из муки и NH4Cl. Во избежание испарения воды, сосуд залит сверху слоем смолы. Работа элемента протекает по схемам:
отрицательный электрод положительный электрод
Zn = Zn•• + 2 e- 2 NH4• + 2 e– = 2 NH4
и затем (вторичные реакции):
2 NH4 = 2 NH3 + 2 H 2 H + MnO2 = H2O + MnO 2 NH3 + 2 H2O = 2 NH4OH
Cухие элементы довольно широко используются в практике.
Аммоний фторид (NH4F) интересен тем, что хорошо растворим во льде (до 10 вес.%).
Аммоний нитрат (аммиачная селитра, NH4NO3) является основой сложных азотных удобрений и служит также для приготовления некоторых взрывчатых смесей. Взрывной распад NH4NO3 протекает в основном по уравнению:
2 NH4NO3 = 4 H2O + N2 + O2 + 238 кДж.
Применяемый в практике взрывных работ аммонал представляет собой тесную смесь нитрата аммония (72%), алюминия в порошке (25%) и угля (3%). Смесь взрывается только от детонации. Наряду с NH4ClO4, он часто вводится в состав твёрдых реактивных топлив. Смешением порошка NH4NO3 c водой (1:1 по массе) может быть создано охлаждение до –13 °С.
Твёрдое реактивное топливо состоит из тщательно гомогенизированной (т.е. приведённой к однородности) смеси окислителя, горючего и добавок специального назначения (способствующих ускорению горения, устойчивости при хранении и т.д.). Его удельный импульс примерно таков же, как у смеси спирта с кислородом. К числу наиболее подходящих окислителей относятся NH4ClO4 и NH4NO3, а горючим обычно служат синтетические полимеры типа каучука и т. п. Такое твёрдое топливо может содержать, например, 75% NH4ClO4, 22% горючего полимера и 3% специальных добавок. В отсутствие горючего термический распад перхлората аммония протекает в основном по уравнению:
4 NH4ClO4 = 6 H2O + 4 HCl + 2 N2 + 5 O2 + 656 кДж.
Сернокислыйаммоний (NH4)2SO4 в больших количествах употребляется сельским хозяйством как азотное удобрение. При нагревании до 357 °С он разлагается с частичным отщеплением аммиака, тогда как чистый NH4HSO4 не только плавится, но и кипит без разложения (т. пл. 251, т. кип. 490 °С). Источником промышленного получения (NH4)2SO4 могут служить газы коксовых печей, наряду с различными другими веществами всегда содержащие аммиак. В среднем из каждой тонны перерабатываемого на кокс каменного угля получается 12 кг сульфата аммония. Последний является значительно более концентрированным азотным удобрением, чем навоз. Так, для внесения в почву 1 кг связанного азота необходимо затратить приблизительно 200 кг навоза или 5 кг (NH4)2SO4.
Кислыйуглекислыйаммоний (гидрокарбонат аммония, NH4HCO3) применяется при хлебопечении (главным образом в кондитерском производстве), что основано на его способности легко разлагаться при нагревании по схеме:
NH4HCO3 = NH3 + H2O + CO2
c выделением газов, придающих тесту необходимую пористость. Сульфидаммония [(NH4)2S] является одним из основных реактивов аналитической химии.
При нагревании соли аммония довольно легко разлагаются. Характер разложения зависит от свойств кислоты, образующей анион. Если последняя является окислителем, происходит окисление аммиака. Если кислота окислителем не является, характер распада определяется её летучестью при температуре разложения. Из солей нелетучих кислот (например, Н3РО4) выделяется только аммиак, если же кислота летуча (например, НСl), то при охлаждении она вновь соединяется с NH3. Результат подобного распада и последующего обратного соединения сводится к тому, что рассматриваемая соль (например, NH4Cl) возгоняется.
Соли аммония при одинаковости структурного типа тем устойчивее по отношению к нагреванию, чем сильнее кислоты, их образующие (если кислота не окислитель). Так, термическая устойчивость солей уменьшается в ряду: HI-HBr-HCl-HF-HSH-HOH.
Давление диссоциации NH4SH превышает 30 мм рт. ст. уже обычных температурах. Но константа кислотной диссоциации HSH (K1 = 1·10-7) несравненно больше, чем у НОН (К1 = 2·10–16). Отсюда следует, что соединения аммиака с водой могут быть в свободном состоянии устойчивы лишь при низких температурах. В этих условиях действительно образуются два кристаллогидрата — 2NH3·H2O и NH3·H2O, по составу отвечающие соответственно оксиду аммония (NH4)2O и его гидроксиду NH4OH. Первое соединение плавится (с разложением) при –78, второе — при –77 °С. Известен также кристаллогидрат NH3·2H2O (т. пл. –97 °С). Кристаллы NH4OH построены на основе Н-связей: d(OH···N) = 278 пм, d(NH···O) = 325 пм и d(OH···O) = 276 пм, тогда как связи NH···N отсутствуют.
Получение хлорида аммония прокаливанием смеси сульфата аммония и хлорида натрия может служить примером транспортных реакций, особенностью которых является образование конечных продуктов в большем или меньшем удалении от исходных. Такие процессы протекают с промежуточным возникновением газовой фазы. Так, в рассматриваемом случае при прокаливании исходной смеси:
(NH4)2SO4 + 2 NaCl = Na2SO4 + 2 NH3 + 2 HCl
и затем (вдали от зоны накаливания)
2 NH3 + 2 HCl = 2 NH4Cl.
Транспортные реакции могут быть иногда с успехом использованы для разделения веществ, их очистки, выращивания кристаллов и т. д.
Попытки выделить радикал аммоний (NH4) в свободном состоянии успеха не имели, так как при обычных условиях он уже в момент образования распадается на аммиак и водород по схеме:
2 NH4 = 2 NH3 + H2.
Всё же при очень низких температурах, благодаря сильному замедлению реакции распада, радикал этот может существовать. Если действовать NH4I на синий раствор металлического натрия в жидком аммиаке, то он обесцвечивается вследствие реакции по схеме:
Na + NH4I = NaI + NH4.
Так как выделения водорода при этом не наблюдается и образовавшаяся бесцветная жидкость легко присоединяет иод по схеме:
2 NH4 + I2 = 2 NH4I,
весьма вероятно, что в ней содержится свободный NH4. Заметное разложение последнего с выделением водорода начинает идти лишь выше –40 °С. Связь между молекулой NH3 и атомом водорода осуществляется только за счёт межмолекулярных сил. Существует предположение, что свободный аммоний является важной составной частью планет Урана и Нептуна.
При действии на соли аммония сильных щелочей происходит выделение аммиака по реакции:
NH4Cl + NaOH = NaCl + NH4OH = NaCl + NH3 + H2O.
Этим можно пользоваться для лабораторного получения аммиака, а также для открытия ионов NH4+ в растворе: к последнему добавляют щёлочи и затем обнаруживают выделившийся аммиак по запаху или действию его на влажную лакмусовую бумажку.
Реакции замещения водорода менее характерны для аммиака, чем реакции присоединения. Однако при высоких температурах он способен замещать свои водороды на металл, например, по реакции:
2 Аl + 2 NH3 = 2 AlN + 3 H2 + 389 кДж.
Прокаливанием металлов в атмосфере аммиака чаще всего и получают нитриды. Последние представляют собой твёрдые вещества, большей частью очень устойчивые по отношению к нагреванию. Нитриды активных металлов легко разлагаются водой с выделением аммиака по схеме:
Mg3N2 + 6 H2O = 3 Mg(OH)2 + 2 NH3.
Нитриды малоактивных металлов по отношению к воде весьма устойчивы.
Нитридыможно получать непосредственно взаимодействием металлов с азотом при нагревании. Так, марганец выше 1200 °С загорается в атмосфере азота и образует Mn3N2. Аммиак может взаимодействовать не только с металлами, но и с оксидами и галогенидами; реакции идут по уравнениям:
3 Сu2O + 2 NH3 = 3 H2O + 2 Cu3N или
CrCl3 + NH3 = 3 HCl + CrN.
Протекание этих процессов обусловлено летучестью воды или галогеноводородов при высоких температурах.
Во многих нитридах видимая по составу валентность металла совместима с её обычными значениями, в других же случаях сама простейшая формула указывает на сложность структуры. Так, из нитридов уже рассмотренных металлов к первому типу относятся Mn3N2, Mn5N2, CrN, MoN, WN, ко второму — Mn6N5, Mn2N, Mn4N, Re3N, Re2N, Cr2N, Mo3N, Mo2N, W2N.
Помимо нитридов продуктами полного замещения водорода аммиака на металл являются и некоторые более сложные производные, например, NMoCl3.
Нитридные производные неметаллов наиболее разнообразны у серы. Важнейшим из них является нитрид состава S4N4, называемый нитридом серы. Он образуется при взаимодействии серы с жидким аммиаком:
16 NH3 + 10 S Ы 6 (NH4)2S + S4N4.
Равновесие этой обратимой реакции может быть смещено вправо прибавлением AgI, связывающего ионы S2- по схеме:
(NH4)2S + 2 AgI = Ag2SЇ + 2 NH4I.
Нитрид серы выделяется из фильтрата при его упаривании. Он может быть очищен возгонкой в вакууме, а также перекристаллизацией из сероуглерода или бензола. Аналогичное производное есть у селена, для теллура известен нитрид состава Te3N4.
Теплота образования нитрида серы -460 кДж/моль. Чистый нитрид серы образуют оранжево-жёлтые кристаллы (т. пл. 179 °С). В воде он нерастворим, но очень медленно разлагается при контакте с ней, в основном по уравнению:
2 S4N4 + 15 H2O = 2 (NH4)2S3O6 + (NH4)2S2O3 + 2 NH3.
Концентрированной иодоводородной кислотой он может быть восстановлен до H2S и NH3 по схеме:
S4N4 + 20 HI = 4 H2S + 4 NH3 + 10 I2.
При нагревании выше температуры плавления (а также при ударе) нитрид серы со взрывом распадается на элементы. Оранжево-красный Se4N4 и жёлтый Te3N4 ещё более взрывчаты.
Термическим разложением паров нитрида серы (при 300 °С под давлением 0,01 мм рт. ст.) может быть получен бесцветный нитрид S2N2 летучий (с запахом иода) и растворимый во многих органических жидкостях. Взрывной распад этого нитрида на элементы происходит при его растиранием или нагреванием до +30 °С. Хранение при более низких температурах сопровождается постепенным образованием S4N4 или нерастворимого в обычных растворителях полимера (-S-N=S=N-)х с большим значением х. Во влажном воздухе получается преимущественно димер, в сухом — полимер. Последний имеет золотисто-жёлтый цвет с металлическим блеском и в спрессованном состоянии обладает полупроводниковыми свойствами.
Нагреванием S4N4 с серой в присутствии СS2 до 110 °С (под давлением) и последующей отгонкой продукта реакции в высоком вакууме может быть получен S4N2 (ранее принималась формула S5N2). Молекула его представляет собой шестичленный цикл типа S(=N–S–)S. Это тёмно-красное вещество плавится при +23 °С, но затвердевает лишь с трудом (так как легко переохлаждается). Оно обладает очень сильным запахом и растворимо во многих органических жидкостях (но не в воде). При обычных условиях S4N2 разлагается лишь медленно, а при 100 °С — со взрывом. Сообщалось также о получении растворимого в CS2 нитрида S11N2 (т. разл. 150 °С), которому приписывается бициклическое строение с общей для двух колец из атомов серы группировкой N–S–N.
При замещении в молекуле аммиака только двух атомов водорода получаются имиды, а при замещении лишь одного — амиды металлов. Первые содержат в своём составе двухвалентный радикал =NH (имино-группа), вторые — одновалентный радикал –NH2 (аминогруппа). Так при пропускании сухого NH3 над расплавленным металлическим натрием по реакции:
2 Na + 2 NH3 = 2 NaNH2 + H2 + 146 кДж
образуется бесцветный кристаллический амид натрия, являющийся типичной солью с анионом NH2-. Синтез хорошо идёт при 350 °С. В расплавленном состоянии амид натрия (т. пл. 206 °С) хорошо проводит электрический ток, а при нагревании разлагается лишь около 500 °С. Водой он тотчас разлагается по уравнению:
NaNH2 + H2O = NH3 + NaOH.
Амид натрия находит применение при органических синтезах.
Из других амидов довольно устойчивы по отношению к нагреванию только производные наиболее активных металлов, тогда как остальные легко разлагаются (иногда со взрывом). Так, Cr(NH2)3 начинает отщеплять аммиак уже при 100 °С.
Растворимые в жидком аммиаке амиды металлов ведут себя как типичные основания. Лучше всех растворимы в жидком аммиаке амиды Cs, Rb и К, тогда как NaNH2 растворим хуже, а производные остальных металлов либо мало растворимы, либо практически нерастворимы.
Имиды металлов мало изучены. Производные наиболее активных металлов могут быть получены осторожным нагреванием их амидов например по схеме:
2 LiNH2 = NH3 + Li2NH,
а некоторых других (например, Ge, Sn) — с помощью реакций в жидком аммиаке. При дальнейшем нагревании имиды металлов либо переходят в соответствующие нитриды, либо полностью разлагаются (иногда со взрывом).
В отличие от металлов имидные производные весьма характерны для серы. Обусловлено это, в частности, двухвалентностью иминогруппы, вследствие чего она валентно-подобна атому серы.
Некоторые из имидов серы со структурой точки зрения производятся непосредственно от молекулы S8 путём замены в ней атомов серы на радикалы NH. Сюда относится прежде всего S7NH (гептасульфуримид), являющейся одним из продуктов химического взаимодействия серы с жидким аммиаком. Водород этого малоустойчивого в обычных условиях бесцветного твёрдого вещества (т. пл. 113 °С) может быть замещён на атомы или радикалы и металлического, и неметаллического характера. Известны, в частности, зелёная соль натрия (NaNS7) и красная сульфо-кислота (S7NSO3H).
Галогениды азота. Наряду с производными металлов известны продукты замещения водородов аммиака на галоген. Примером может служить хлористый азот (NCl3), характеризующийся следующими параметрами: d(NCl) = 176 пм, РClNCl = 107°. Пары этого вещества (т. пл. -27, т. кип. 71 °С) обладают резким запахом и сильно действуют на слизистые оболочки. Растворы его в некоторых органических растворителях при отсутствии света сохраняются без изменения довольно долго. В воде он почти нерастворим, но медленно разлагается ею на NH3 и HOCl. Его получают в виде маслянистых капель при действии хлора на крепкий раствор хлористого аммония:
NH4Cl + 3 CI2 = 4 HCl + NCl3.
Уже при нагревании выше 90 °С (или ударе) хлористый азот со взрывом распадается на элементы. Термический распад идёт по уравнению:
2 NCl3 = N2 + 3 Cl2 + 460 кДж.
Аналогичное бромистое производное образуется в результате взаимодействия раствора аммиака с избытком брома (лучшие условия — Br2 : NH3 = 2,5 и рН = 4,5). Действием паров брома на избыток аммиака под уменьшенным давлением с последующим охлаждением продуктов реакции до –75 °С может быть получено тёмно-красное вещество состава NBr3·6NH3, разлагающееся со взрывом уже при –70 °С.
При действии иода на крепкий раствор NH3 выделяется тёмно-коричневый осадок, так называемого йодистого азота, представляющий собой в действительности соединение NI3 с переменным количеством аммиака (или продукт неполного замещения водорода последнего на иод). Медно-красные игольчатые кристаллы состава NI3·NH3 были выделены и в индивидуальном состоянии. Они взрываются уже при нагревании выше 26 °С. Содержащее азот и иод в атомном отношении 1:3 вещество может быть получено, по-видимому, действием газообразного аммиака на двойное соединение KBr·IBr по уравнению:
3 (KBr·IBr) + 4 NH3 = 3 KВr + 3 NH4Br + NI3.
После промывания продуктов реакции водой остаётся чёрный осадок NI3. Йодистый азот крайне неустойчив и в сухом виде взрывается от малейшего прикосновения.
В противоположность другим галогенидам фтористый азот (NF3) является соединением экзотермическим (теплота образования 125 кДж/моль) и невзрывчатым. Он может быть получен электролизом расплавленного аммоний-гидродифторида (NH4HF2) и образуется также при взаимодействии аммиака с фтором:
4 NH3 + 3 F2 = 3 NH4F + NF3.
Однако без сильного разбавления реакционной смеси азотом реакция эта протекает настолько энергично, что большая часть первоначальных продуктов распадается на N2 и HF.
Молекула NF3 пирамидальна d(NF) = 137 пм, РFNF = 102°. Её малая полярность обусловлена, по-видимому, обратным направлением диполей 3F¬N и N®2e к свободной электронной паре атома азота.
Фтористый азот представляет собой бесцветный газ (т. пл. –209, т. кип. –129 °С). По отношению к нагреванию и различным химическим воздействиям он весьма устойчив. Так, при обычных условиях NF3 не реагирует с сухим стеклом, ртутью, водой и даже КОН. Он ядовит. В воде NF3 почти нерастворим, а взаимодействие его с водяным паром начинается лишь под воздействием электрической искры и медленно протекает по схеме:
2 NF3 + 3 H2O = 6 HF + N2O3.
Реакция с водородом в тех же условиях сопровождается взрывом, причём продуктами её являются HF и N2.
Взаимодействие NF3 c F2 и SbF3 или AsF3 (нагреванием под давлением или в тихом разряде при –78 °С) были получены фтористые аналоги солей аммония — [NF4]SbF6 и [NF4]AsF6. Первое из этих гигроскопичных солеобразных веществ разлагается лишь около 300 °С, второе — при 270 °С. С водой они гладко взаимодействуют по схеме:
2 NF4ЭF6 + 14 H2O = 2 NF3 + O2 + 2 HЭ(OH)6 + 14 HF.
С точки зрения устойчивости и реакционной способности соотношение между NF3 и NCI3 аналогично соотношению между F2O и Cl2O: и в том, и в другом случае хлористые производные взрывчаты, тогда как фтористые устойчивы и химически гораздо более инертны.
Из продуктов неполного замещения водорода аммиака на галоген сравнительно хорошо изучен только хлорамин (NH2Cl), представляющий собой бесцветную маслянистую жидкость с резким запахом (т. пл. –60 °С). Получают его действием NaOCl на взятое по расчёту количество аммиака, после чего жидкость подвергают перегонке в вакууме и дистиллят обрабатывают безводным К2СО3 (для связывания воды). Получить NH2Cl можно и по реакции:
2 NH3 + Cl2 = NH4Cl + NH2Cl,
если исходные газы достаточно разбавлены азотом. По строению молекулы хлорамин подобен аммиаку. Он хорошо растворим в воде, но его основные свойства выражены у него крайне слабо (К = 1·10-15). Несколько сильнее выражены кислотные свойства хлорамина.
В растворе NH2Cl подвергается гидролизу по схеме:
NH2Cl + H2O Ы NH3 + HOCl,
чем и обусловлены характерные для него окислительные свойства. При хранении водных растворов происходит постепенное разложение хлорамина вследствие различных вторичных реакций. Некоторые его органические производные применяются в медицине, для стерилизации воды и дегазации (уничтожения отравляющих веществ).
Продуктом замещения одного из водородов аммиака на гидроксильную группу является гидроксиламин (NH2OH). Он образуется при электролизе азотной кислоты (с ртутным или свинцовым катодом) в результате восстановления HNO3 по схеме:
HNO3 + 6 H ® 2 H2O + NH2OH.
Гидроксиламин представляет собой бесцветные кристаллы. Используется он главным образом как сильный восстановитель (окисляется до N2 или N2O).
Под уменьшенным давлением гидроксиламин (т. пл. 33 °С) может быть перегнан без разложения (т. кип. 58 °С при 22 мм рт. ст.), тогда как при нагревании выше 100 °С он разлагается (часто со взрывом). Постепенно разлагается он и при обычных условиях.
Расплавленный NH2OH является хорошим растворителем некоторых солей. С водой он образует гидрат гидроксиламина (NH2OH·H2O), характеризующийся слабо выраженными основными свойствами (К = 2·10-8). В сочетании с дымящей HNO3 соединение это иногда используется как реактивное топливо. С кислотами гидроксиламин даёт соли, из которых легкорастворимая хлористая — NH2OH·HCl (т. пл. 151 °С) — является обычным препаратом, поступающим в продажу, а малорастворимая фосфорнокислая соль (3NH2OH·H3PO4) может быть использована (путём нагревания её под уменьшенным давлением) для получения безводного NH2OH.
Разбавленные водные растворы солей гидроксиламина довольно устойчивы, тогда как крепкие быстро разлагаются (особенно в присутствии щелочей) с образованием NH3, N2 и N2O. Такой распад сильно ускоряется в присутствии платиновой черни. Окислители обычно переводят гидроксиламин либо в N2O, либо в N2, например по реакции:
6 NH2OH + 4 HNO3 = 3 N2O + 4 NO + 11 H2O или
2 NH2OH + HOCl = N2 + HCl + 3 H2O.
Для гидроксиламина довольно характерна и окислительная функция. Например, он способен окислять Fe(OH)2 до Fe(OH)3, H2SO3 до H2SO4 и т. д. Эта окислительная функция более выражена в кислой среде, тогда как наиболее характерная для гидроксиламина восстановительная — в щелочной. Иногда изменение характера среды полностью меняет поведение гидроксиламина. Так, в уксуснокислой среде он восстанавливает I2 до HI, а в сильно солянокислой — окисляет HI до I2.
Подобно замещению водорода, реакции окисления для аммиака сравнительно малохарактерны. На воздухе он не горит, но, подожжённый в атмосфере кислорода, сгорает жёлтым пламенем с образованием азота и водяного пара:
4 NH3 + 3 O2 = 6 H2O + 2 N2 + 1267 кДж.
Хлор и бром энергично реагирует с аммиаком по схеме:
2 NH3 + 3 Г2 = 6 НГ + N2
Так же окисляют они аммиак и в растворе. По отношению к большинству других окислителей NH3 при обычных условиях устойчив.
Наиболее важным продуктом частичного окисления аммиака является гидразин (N2H4), образующийся по реакции:
2 NH3 + NaOCl = H2O + N2H4 + NaCl.
Хороший выход гидразина при его получении может быть достигнут в присутствии некоторых органических веществ. Обычно в реакционную смесь вводят 0,2% желатина. Сама реакция идёт в две стадии по уравнениям:
NH3 + NaOCl = NaOH + NH2Cl (быстрая реакция) и
NH2Cl + NH3 + NaOH = H2O + NaCl + N2H4 (медленная реакция),
причём роль желатина сводится к предотвращению вредной побочной реакции
2 NH2Cl + N2H4 = N2 + 2 NH4Cl.
Такой ход процесса подтверждает возможность получения солянокислого гидразана прямым взаимодействием хлорамина с аммиаком:
NH2Cl + NH3 = N2H4·HCl.
Гидразин частично образуется также при освещении струи аммиака лучами ртутной кварцевой лампы.
Как видно из уравнения, под действием окислителя каждая молекула аммиака теряет в данном случае один атом водорода, причём оставшиеся радикалы NH2 соединяются друг с другом. Структурная формула гидразина будет, следовательно, H2N–NH2.
Гидразин представляет собой бесцветную жидкость, дымящую на воздухе и легко смешивающуюся с водой. Он находит применение в качестве сильного восстановителя (окисляется до N2).
Свободный гидразин (т. пл. 2, т. кип. 113 °С) способен присоединять молекулу воды, давая гидрат гидразина — N2H4·H2O. Последний представляет собой бесцветную жидкость (т. пл. –52, т. кип. 119 °С) и является слабым основанием (К1 = 1·10-6). Присоединение второй молекулы воды идёт уже с трудом, и отвечающая ему константа диссоциации очень мала (К2 = 9·10–16). Присоединяя молекулы кислот, гидразин может образовывать два ряда солей; например, N2H4·HCl и N2H4·2HCl. В продажу обычно поступает малорастворимый сульфат N2H4·H2SO4 (т. пл. 254 °С с разл.).
В отличие от аммиака, гидразин является эндотермическим соединением (теплота образования из элементов –96 кДж/моль). Пары его способны сгорать фиолетовым пламенем по реакции:
N2H4 + O2 = 2 H2O + N2 + 581 кДж.
На этом основано использование гидразина в качестве реактивного топлива. Его удельный импульс в комбинации с кислородом доходит до 270, с озоном — до 280, а с фтором — до 300 с (т.е. гидразин несколько эффективнее аммиака). Ещё более эффективны метилзамещённые гидразины. Так, (СН3)2NNH2 способен дать с кислородом удельный импульс 310 с.
Жидкий гидразин характеризуется высоким значением диэлектрической проницаемости (e = 53 при 20 °С) и является хорошим ионизирующим растворителем для ряда солей. Его собственная электролитическая диссоциация невелика: [N2H5+][N2H3-] = 2·10–25. С металлическим натрием гидразин взаимодействует по схеме:
2 Na + 2 N2H4 = 2 NaN2H3 + H2.
Образующийся гидразинид натрия представляет собой весьма взрывчатое твёрдое вещество жёлтого цвета, хорошо растворимое в избытке гидразина.
В водных растворах гидразин восстанавливает иод до йодистого водорода, соли серебра и ртути — до металлов, соли меди — до её закиси и т. д. Сам он при этом окисляется до свободного азота, но основной процесс обычно осложняется побочными реакциями. Полностью до N2 гидразин может быть окислен лишь в строго определённых реакциях (например, иодом при рН = 7 ё 7,2). Интересно, что его практически нерастворимое в воде двойное соединение с хлоридом хрома (II) состава CrCl2·2N2H4 очень устойчиво к действию окислителей, хотя обе его составные части являются восстановителями.
Окислительная функция у гидразина почти отсутствует, но действием очень сильных восстановителей (водорода в момент выделения, Sn••, Ti•••) он всё же может быть восстановлен до аммиака. В форме разбавленного водного раствора гидразин является хорошей антикоррозионной добавкой к воде, идущей для питания паровых котлов (так как освобождает её от растворённого кислорода и одновременно сообщает ей слабощелочную реакцию). И сам гидразин, и его производные ядовиты.
Взаимодействием NF3 при нагревании с углём (в кипящем слое) по схеме:
4 NF3 + C = CF4 + 2 N2F4
может быть получен фтористый аналог N2H4 — тетрафторгидразин. Он газообразен (т. пл. –162, т. кип. –73 °С), но его критическая температура лежит сравнительно высоко (+36 °С). И в газообразном, и в жидком состоянии он частично диссоциирован на свободные радикалы по схеме:
N2F4Ы 2 NF2 (при 25 °С степень диссоциации составляет 0,02%).
Радикал NF2, несмотря на наличие “холостого” электрона, обладает сравнительно высокой устойчивостью. Тетрафторгидразин был предложен в качестве возможного окислителя реактивного топлива.
При взаимодействии гидразина с азотистой кислотой по схеме:
N2H4 + HNO2 = 2 H2O + HN3
образуется азотистоводороднаякислота (H–N=NєN), представляющая собой бесцветную летучую жидкость с резким запахом. Для успешного протекания реакции требуется достаточно кислая среда. Практически кислоту и её соли удобнее получать, исходя из азида натрия, который образуется в результате протекающей при 190 °С реакции по схеме:
NaNH2 + N2O = H2O + NaN3.
По силе азотистоводородная кислота близка к уксусной, а по растворимости солей (азидов) похожа на соляную. Подобно самой HN3, некоторые азиды при нагревании или ударе сильно взрываются. На этом основано применение азида свинца [Pb(N3)2] в качестве детонатора, т. е. вещества, взрыв которого вызывает мгновенное разложение других взрывчатых веществ.
Ион N3- имеет линейное строение. Кислотная функция HN3 (т. пл. –80, т. кип. +37 °С) характеризуется значением К = 2·10–5. При нагревании паров HN3 выше 300 °С они с сильным взрывом разлагаются, в основном по реакции:
2 HN3 = H2 + 3 N2 + 589 кДж.
В безводном состоянии азотистоводородная кислота способна взрываться не только при нагревании, но и просто от сотрясения сосуда. Напротив, в достаточно разбавленном водном растворе она практически устойчива, так как реакция её разложения по уравнению
HN3 + H2O = N2 + NH2OH
идёт крайне медленно. Пары HN3 очень ядовиты, а её водные растворы вызывают воспаление кожи.
Помимо кислотной функции для HN3 характерна также окислительная. Взаимодействие её с HI сопровождается выделением иода и образованием продуктов восстановления азотистоводородной кислоты — N2 и NH3. Смесь HN3 с крепкой HCl при нагревании растворяет золото и платину, т. е. ведёт себя аналогично царской водке. При действии HN3 на металлы происходит образование не только соответствующих азидов, но и N2 и NH3, тогда как свободный водород не выделяется. По всем этим реакциям азотистоводородная кислота похожа на азотную. Основной причиной этого сходства является наличие в молекулах обоих соединений азота (+5).
Восстановительная функция для HN3 не характерна, но с некоторыми сильными окислителями она всё же взаимодействует. Так, азотистая кислота окисляет HN3 по уравнению:
HNO2 + HN3 = N2 + N2O + H2O.
Реакция эта может быть использована для количественного анализа азидов.
Соли HN3, как правило, бесцветны. Производные некоторых наиболее активных металлов могут быть расплавлены без разложения, и распад их на металл и азот происходит только при несколько более сильном нагревании. Например, KN3 плавится при 343 °С, а разлагается при 355 °С. Азид свинца (ПР = 2•10–9) взрывается при 327 °С и от удара.
Известны так же продукты замещения водорода на галоген. Фторазид (FN3) образуется при взаимодействии HN3 и F2 в токе азота по уравнению:
4 HN3 + 2 F2 = NH4F + N2 + 3 FN3.
Это зелёный газ (т. пл. –152, т. кип. –82 °С), медленно разлагающийся по схеме:
2 FN3 = 2 N2 + N2F2.
Соединение состава N2F2 — дифтордиазин — образуется в качестве одного из продуктов при электролизе аммоний-гидродифторида или действии фтора на натрийазид. Более прямым путём его получения является взаимодействие фторимина с очень тщательно высушенным калийфторидом по уравнению:
2 KF + 2 HNF2 = 2 KHF2 + N2F2.
Получающийся почти со 100%-ным выходом бесцветный газ (похожий по запаху на NO2) малоустойчив и медленно разлагается на N2 и F2 уже при обычных условиях. Тем не менее он может быть разделён на две фракции, образованные цис- и транс-формами молекул F–N=N–F. Несколько более устойчива цис-форма (теплота образования её из элементов –67 кДж/моль, т. пл. –195, т. кип. –106 °С), характеризующаяся параметрами: d(NN) = 121, d(NF) = 138 пм, РNNF = 114°. Для транс-формы (теплота образования из элементов –79,5 кДж/моль, т. пл. –172, т. кип. 111 °С) параметры: d(NN) = 123, d(NF) = 140 пм, РNNF = 106°.
Хлоразид (ClN3) получается при взаимодействии азотистоводородной и хлорноватистой кислот по схеме:
ClOH + HN3Ы H2O + ClN3.
В кислой среде реакция протекает слева направо, в щелочной — справа налево. Хлорозид бесцветный газ (т. пл. –100, т. кип. –15 °С), соответствующее ему бромистое производное — жидкость красного цвета (т. пл. –45 °С). Желтоватые кристаллы иодазида могут быть получены взаимодействием азида серебра с иодом по реакции:
I2 + AgN3 = AgI + IN3.
Все галогеназиды чрезвычайно взрывчаты. Водой они постепенно разлагаются гидролитически.
Взаимодействием ClN3 (где хлор поляризован положительно) с хлоридами некоторых металлов (где хлор поляризован отрицательно) могут быть получены их смешанные азидохлориды, например, по схеме:
WCl6 + ClN3 = Cl2 + WCl5N3.
Разложение этого азидхлорида идёт с образованием нитридхлорида по уравнению:
Cl5WN3 = N2 + Cl3WN + Cl2.
Среди продуктов распада HN3 под действием электрического разряда был обнаружен диимид (H–N=N–H), вероятно образующийся по схемам:
HN3® N2 + NH и NH3 + NH ® N2 + N2H2.
Вещество это не выделено.
Аналогичный N2H2 по составу полиимид — (NH)x осаждается при охлаждении жидким азотом продуктов термического разложения HN3 около 1000 °С (по схеме: HN3 ® N2 + NH. Это нерастворимое в воде синее вещество уже при –125 °С переходит в NH4N3.
Кислородные соединения азота.
Для азота известны оксиды, по составу формально отвечающие всем его валентностям от единицы до пяти. Их формулы и названия сопоставлены ниже: N2O NO N2O3 NO2 N2O5
гемиоксид монооксид сесквиоксид диоксид гемипентаоксид.
Азотный ангидрид представляет собой твёрдое вещество, а остальные оксиды при обычных условиях газообразны.
При взаимодействии с раскалённой медью все оксиды азота разлагаются, образуя CuO и N2. По количеству оксида меди и объёму выделившегося азота может быть установлена формула исходного оксида.
За исключением N2O (“веселящего газа”), все оксиды азота ядовиты. Опасность отравления ими усугубляется тем, что дыхательные пути в данном случае раздражаются сравнительно слабо, и тяжёлые явления (боль в груди, сильная одышка и др.) наступают обычно лишь спустя несколько часов после вдыхания газа. Воздух, содержащий 0,5 мг/л окислов азота, при вдыхании его в течение часа может вызвать опасное для жизни заболевание. В качестве мер первой помощи при острых отравлениях рекомендуются обильные приёмы молока, кислородное дыхание, а также впрыскивание камфары. Пострадавший должен находиться в полном покое. При хронических отравлениях наблюдается сердцебиение, катар дыхательных путей, кровохаркание и разрушение зубов. Максимально допустимое содержание оксидов азота в воздухе производственных помещений составляет 0,005 мг/л.
Гемиоксид азота может быть получен разложением азотнокислого аммония, протекающим около 250 °С по уравнению:
NH4NO3 = 2 H2O + N2O + 40 кДж.
Структура молекулы N2О соответствует формуле NєN=O. Гемиоксид азота представляет собой бесцветный газ со слабым приятным запахом и сладковатым вкусом. В воде он довольно хорошо растворим, но химически с ней не взаимодействует.
Выше 500 °С гемиоксид азота разлагается по реакции:
2 N2O = 2 N2 + O2 + 163 кДж.
Поэтому при повышенных температурах он действует как сильный окислитель. Так, тлеющая лучинка в нём вспыхивает. Параллельно приведённой выше реакции термического распада незначительно протекает и побочная:
2 N2O = N2 + 2 NO.
С кислородом N2O не соединяется, а смеси его с водородом и аммиаком при нагревании взрывается. В кислых растворах H2SO3 медленно восстанавливает N2O до свободного азота, Sn•• — до гидроксиламина, а Ti•••— до аммиака.
Вдыхание гемиоксида азота в смеси с воздухом вызывает характерное состояние опъянения, сопровождающееся ослаблением болевых ощущений. На этом основано использование N2O при операциях в качестве наркотика.
Слишком быстрое нагревание NH4NO3 (т. пл. 170 °С) может сопровождаться взрывом. По той же причине нельзя допускать нагрев его расплава выше 300 °С. Параллельно с ведущим к образованию N2O основным процессом частично протекают реакции по схемам:
NH4NO3 = NH3 + HNO3 и 5 NH4NO3 = 9 H2O + 4 N2 + 2 HNO3,
сопровождающиеся дальнейшим разложением азотной кислоты. Поэтому получаемый термическим разложением NH4NO3 [или смеси 2NaNO3+(NH4)2SO4] гемиоксид азота всегда содержит примеси NO и NO2, от которых может быть освобождён пропусканием сквозь раствор FeSO4. Удобным методом получения чистого гемиоксида является слабое нагревание сульфаминовой кислоты с предварительно прокипячённой (для удаления следов оксидов азота) 73%-ной азотной кислотой. Реакция в этих условиях количественно идёт по уравнению:
HNO3 + NH2SO2OH = N2O + H2SO4 + H2O.
Из 4 г NH2SO2OH и 10 см3 HNO3 может быть получено около 1 л N2O.
Молекула N2O линейна [d(NN) = 113, d(NO) = 118 пм] и малополярна. Гемиоксид (т. пл. –91, т. кип. –89 °С) является постоянной составной частью воздуха (5·10-5 объёмн.%). Критическая температура этого газа равна +36 °С при критическом давлении 72 атм. Один объём воды поглощает при 0 °С около 1,3, а при 25 °С — 0,6 объёма N2O. В результате охлаждения насыщенных растворов образуется кристаллогидрат N2O·6H2O, нагревание которого может служить методом получения очень чистой N2O. Для наркоза обычно применяют смесь 80% гемиоксида азота с 20% кислорода.
Энергия активации термического распада гемиоксида азота в газовой фазе равна 242 кДж/моль, на Pt она снижается до 138 кДж/моль, а на Au — до 121 кДж/моль.
Образование монооксида азота из элементов при обычных условиях не происходит. Лишь примерно с 1200 °С начинает заметно протекать обратимая реакция:
N2 + O2 + 180 кДж Ы 2 NO.
Около 1500 °С равновесие ещё почти нацело смещено влево. Устанавливается оно при этих условиях чрезвычайно медленно: для достижения равновесного состояния требуется 30 ч. Напротив, более высоким температурам отвечает не только большее содержание NO в газовой фазе, но и несравненно более быстрое достижение равновесия, которое при 3000 °С устанавливается практически мгновенно. По этим причинам NO всегда образуется в атмосфере при грозовых разрядах.
Несмотря на эндотермичность монооксида азота, при обычных условиях он вполне устойчива. В лаборатории его чаще всего получают по реакции:
3 Cu + 8 HNO3 = 3 Cu(NO3)2 + 2 NO + 4 H2O.
Это уравнение отражает лишь главное направление процесса. На самом деле одновременно протекают и побочные реакции, в результате чего к монооксиду азота оказываются примешенными другие газообразные продукты — NO2, N2O, и N2. Содержание этих примесей зависит от концентрации исходной кислоты и прочих условий опыта.
Монооксид азота представляет собой бесцветный газ, сравнительно малорастворимый в воде и химически с ней не взаимодействующий. Свой кислород он отдаёт лишь с трудом. Поэтому горящая лучина в атмосфере NO гаснет.
Очень чистый монооксид азота может быть получен пропусканием сернистого газа в тёплую азотную кислоту (плотность 1,15 г/см3). Равномерную струю NO можно получить по реакции:
FeCl2 + NaNO2 + 2 HCl = FeCl3 + NaCl + H2O + NO,
медленно приливая крепкий раствор NaNO2 в колбу, содержащую солянокислый раствор FeCl2 (или FeSO4). Ещё один удобный метод получения NO основан на реакции:
2 HNO2 + 2 HI = 2 NO + I2 + 2 H2O.
Для этого 50%-ная серная кислота медленно добавляется к раствору 4 М относительно NaNO2 и 1 М относительно КI.
Образование монооксида азота из элементов является цепной реакцией, развивающейся по схеме:
O + N2® NO + N N + O2® NO + O и т. д.
Выше 3000 °С содержание NO в равновесных смесях начинает снижаться, что обусловлено главным образом диссоциацией молекул О2 на атомы.
Молекула NO характеризуется d(NO) = 115 пм. NO+ является основным ионом на сравнительно небольших высотах верхней атмосферы (примерно до 200 км).
Критическая температура монооксида азота –94 °С при критическом давлении 65 атм. В жидком и твёрдом состояниях NO (т. пл. –164, т. кип. –151 °С) имеет синий цвет. Сто объёмов воды растворяют при 0 °С около 7 объёмов NO. Слабо горящий фосфор гаснет в этом газе, но сильно горящий продолжает гореть.
Смесь NO с равным объёмом Н2 при нагревании взрывается. Под высоким давлением NO (500 атм) он уже при обычных температурах окисляет сернистый газ по уравнению:
2 NO + 2 SO2 = 2 SO3 + N2.
C гидроксидами щелочных металлов NO также при обычных температурах взаимодействует по следующим параллельным реакциям:
4 NO + 2 ЭОН = N2O + 2 ЭNO2 + H2O и 6 NO + 4 ЭОН = N2 + 4 ЭОН + 2 Н2О.
По ряду Li®Cs скорости этих процессов возрастает.
В растворе SO2 восстанавливает NO до N2O, ион Сr•• в кислой среде — до гидроксиламина, а в нейтральной — даже до аммиака. Точно так же до аммиака восстанавливается NO и водородом в момент выделения. Напротив, действием сильных окислителей (CrO3, HMnO4, HOCl и т.п.) NO окисляется до азотной кислоты. Озон легко переводит NO в N2O5. С хлористым водородом NO образует устойчивый лишь в твёрдом состоянии ниже –130 °С красный продукт присоединения состава NO·HСl.
Наиболее характерны для NO реакции присоединения. Так, при взаимодействии его с хлором по реакции:
2 NO + Cl2 = 2 NOCl + 75 кДж
образуется хлористый нитрозил (Сl–N=O), представляющий собой жёлтый газ. Непосредственно соединяется NO и с кислородом. Известен также ряд комплексных соединений, содержащих NO во внутренней сфере.
Спокойно протекающая реакция соединения NO c кислородом воздуха ведёт к образованию диоксида азота по уравнению:
2 NO + O2 = 2 NO2 + 113 кДж.
Диоксид азота представляет собой бурый газ, легко сгущающийся в жидкость, кипящую при +21 °С. Будучи охлаждена ниже –14 °С, жидкость эта застывает в бесцветную кристаллическую массу. Определение молекулярного веса в газообразном состоянии даёт цифры, лежащие между простым (46) и удвоенным (92) его значениями, причём цифры эти изменяются в зависимости от температуры опыта, уменьшаясь при её повышении и увеличиваясь при понижении.
Такие результаты обусловлены наличием равновесия между молекулами диоксида азота (NO2) и димера (N2O4). Определение молекулярного веса около 140 °С показывает, что при этих условиях в газе имеются только молекулы диоксида азота, тогда как при более низких температурах они частично соединяются попарно, образуя молекулы N2O4. Так как процесс образования из нейтральных молекул одного и того же вещества более сложных частиц с удвоенным, утроенным и т. д. молекулярным весом называется полимеризацией, можно сказать, что при температуре ниже 140 °С NO2 частично полимеризуется (точнее — димеризуется) в N2O4. Это происходит тем в большей степени, чем ниже температура, и вблизи точки замерзания (–11 °С) вещество состоит уже исключительно из молекул N2O4. Напротив, при нагревании димер диссоциирует на простые молекулы.
Каждой промежуточной между –11 °С и +140 °С температуре отвечает определённое равновесие обратимой реакции:
2 NO2Ы N2O4 + 59 кДж.
Так как димер бесцветен, а диоксид имеет красно-бурый цвет, за смещением равновесия при нагревании или охлаждении газовой смеси легко следить по изменению её окраски.
Склонность молекул О=N=O к взаимодействию друг с другом обусловлена наличием в каждой из них одного непарного электрона (при атоме азота). Сочетание двух таких электронов и создаёт связь N–N в молекуле N2O4. Неустойчивость последней является следствием непрочности этой связи: d(NO) = 119, d(NN) = 176 пм, Р ОNО = 135°.
Диоксид азота является очень сильным окислителем. Уголь, сера, фосфор и т. д. легко сгорают в нём. С парами многих органических веществ он даёт взрывчатые смеси. Склонность к реакциям присоединения выражена у диоксида значительно слабее, чем у монооксида.
Лабораторное получение NO2 и N2O4 удобно вести прокаливанием сухого Pb(NO3)2 (в смеси с равным объёмом предварительно прокалённого песка). Выделяющийся при разложении по схеме:
2 Рb(NO3)2 = 2 PbO + 4 NO2 + O2
диоксид азота собирают в охлаждаемом приёмнике.
В жидком диоксиде азота имеет место очень незначительная электролитическая диссоциация по схеме:
NO2 + NO2Ы NO++ NO3-.
Металлический натрий реагирует с ним быстро, но спокойно, образуя NO и NaNO3, который, как и другие соли, в жидком диоксиде нерастворим. Электропроводность твёрдого диоксида азота в 1000 раз больше, чем жидкого. Димер диоксида находит применение в реактивной технике и может быть использован как теплоноситель.
Если подсчитать общее число внешних электронов в молекуле NO2, то получится цифра 17 (их 5 у азота и 2·6 у кислорода). Так как валентная связь осуществляется электронной парой, последняя должна быть системой более устойчивой, чем неспаренный электрон. Можно поэтому ожидать, что молекулы с нечётным числом электронов (“нечётные” молекулы) будут склонны к димеризации (т.е. попранному сочетанию). Правильность этого предположения подтверждается тем обстоятельством, что подавляющее большинство всех способных к устойчивому существованию веществ состоит из “чётных” молекул. К очень немногочисленным исключениям относится монооксид азота, который имеет в молекуле 11 внешних электронов и легко соединяется с рядом различных веществ, но проявляет заметные признаки димеризации по схеме:
NO + NO Ы N2O2
лишь при низких температурах. В жидком состоянии при –163 °С содержание молекул N2O2 достигает 95%. Твёрдый монооксид азота состоит уже из димерных молекул (NO)2, энергия связи между которыми равна лишь 12,5 кДж/моль. Димеры могут существовать в различных формах, причём относительно более устойчив цис-изомер ONNO, для которого: d(NN) = 175 пм и РNNO = 90°. В жидком или сжатом состоянии (а также при длительном контакте с водой) димерный монооксид медленно разлагается по схеме:
2 N2O2 = N2O + N2O3.
Так как молекула NO содержит изолированный (непарный) электрон, он способен соединяться со свободными радикалами. Это нередко используется для выяснения, является ли тот или иной химический процесс развивающийся по радикальному механизму, цепной реакцией: в таком случае добавление монооксида азота ведёт к обрыву цепей и тем самым к резкому замедлению процесса.
Реакция присоединения к NO кислорода может быть использована для его открытия (в смесях с N2O и др.). Она особенно интересна тем, что является одним из очень немногих известных случаев, когда при повышении температуры химический процесс не только ускоряется, но даже несколько замедляется (средний температурный коэффициент скорости равен 0,9). Объяснение этой аномалии скорости исходит из того, что в реакцию вступают лишь димерные молекулы N2O2, вероятность возникновения которых с повышением температуры очень быстро уменьшается.
Взаимодействие NO2 с NO по обратимой реакции
NO2 + NO Ы N2O3 + 42 кДж
ведёт к частичному образованию сесквиоксида азота (N2O3), который при охлаждении может быть получен в виде синей жидкости. В обычных условиях он неустойчив, и равновесие сильно смещено влево.
Получить сесквиоксид азота (т. пл. –101 °С) удобнее всего, пуская по каплям 50%-ную HNO3 на As2O3 (или крахмал). Образующиеся по реакции:
2 HNO3 + As2O3 = 2 HAsO3 + NO + NO2
в эквивалентных количествах NO и NO2 при пропускании сквозь помещённую в охладительную смесь трубку легко соединяются. Азотистый ангидрид образуется также (в виде голубого порошка) при пропускании электрических искр сквозь жидкий воздух.
Термическая диссоциация азотистого ангидрида начинает идти уже ниже 0 °С. При 25 °С и обычном давлении содержание N2O3 в равновесной системе
N2O3Ы NO2 + NO
составляет лишь 10,5%, при 50 °С — 5,8%, а при 100 °С — 1,2 %. Молекула сесквиоксида азота плоская и имеет несимметричное строение ON—NO2 с параметрами: d(ON) = 114, d(NN) = 186, d(NO) = 121 пм, РONN = 105°, РNNO = 113 и117°. С водой жидкий N2O3 полностью смешивается лишь выше 55 °С (под давлением).
При взаимодействии N2O3 с сильно охлаждённым жидким аммиаком по реакции:
2 NH3 + N2O3 = H2O + 2 NH2NO
образуется оранжево-красный нитрозамид. Он крайне неустойчив и при испарении избытка аммиака разлагается по схеме:
2 NH2NO = N2 + NH4NO2,
но продукты замещения в нём водородов на некоторые органические радикалы известны и в свободном состоянии.
Растворение NO2 (или N2O4) в воде сопровождается образованием азотной (HNO3) и азотистой (HNO2) кислот:
N2O4 + H2O = HNO3 + HNO2.
Тогда как азотная кислота в растворе устойчива, азотистая распадается по обратимой реакции:
2 HNO2Ы H2O + N2O3Ы H2O + NO2 + NO.
Поэтому взаимодействие NO2 с водой практически идёт по уравнению:
3 NO2 + H2O = 2 HNO3 + NO.
Если растворение диоксида азота вести в присутствии избытка кислорода (воздуха), то выделяющаяся NO окисляется им до NO2. При этих условиях можно полностью перевести NO2 в азотную кислоту по суммарной схеме:
4 NO2 + 2 H2O + O2 = 4 HNO3.
Подобным же образом (с образованием солей HNO3) протекает растворение NO2 в щелочах при наличии избытка кислорода. Напротив, в отсутствии последнего по реакции, например:
2 NO2 + 2 NaOH = NaNO2 + NaNO3 + H2O.
образуются соли азотной и азотистой кислот (в отличие от самой HNO2, соли её устойчивы).
Соли азотистой кислоты — нитриты — бесцветны, почти все хорошо растворимы в воде (хуже других — AgNO2). Чаще всего встречается в практике NaNO2, который получают обычно по схеме:
NO2 + NO + 2 NaOH = 2 NaNO2 + H2O.
Соль эта используется при производстве органических красителей.
Лишь немногие нитриды плавятся без разложения. В растворах они постепенно окисляются кислородом воздуха с образованием соответствующих нитратов. Нитрит натрия (т. пл. 283 °С) находит медицинское использование как сосудорасширяющее средство. В больших дозах соли азотистой кислоты весьма ядовиты.
Сама азотистая кислота известна только в разбавленных водных растворах. По силе она лишь немного превышает уксусную кислоту. Наиболее характерны для неё сильно выраженные окислительные свойства, причём восстанавливается она в большинстве случаев до NO. С другой стороны, действием сильных окислителей азотистая кислота может быть окислена до азотной. Типичные примеры характерных для HNO2 окислительно-восстановительных процессов приводят ниже:
2 HNO2 + 2 HI = I2 + 2 NO + 2 H2O
2 HMnO4 + 5 HNO2 = 2 Mn(NO3)2 + HNO3 + 3 H2O
Обе эти реакции протекают в кислой среде.
Скорость окисления азотистой кислотой HI с ростом рН среды уменьшается, а при рН > 4,8 оно прекращается. Параллельно с основной реакцией (восстановлением HNO2 до NO) может частично протекать и побочная:
2 HNO2 + 4 HI = 2 I2 + N2O + 3 H2O.
Ионами Fe•• азотистая кислота восстанавливается до NO, а ионами Sn••— до N2O.
Окисление HNO2 до азотной кислоты посредством H2O2 происходит только в кислой среде и идёт через промежуточное образование надазотистой кислоты — HOONO.
Диссоциация HNO2 по основному типу очень мала и заметно выявляется только в сильнокислых средах. Для равновесия реакции
Н•+ HNO2Ы H2O + NO•
найдено значение К = 2·10-7. В менее кислых средах на неё налагается сильно смещённое вправо равновесие
NO• + NO2’Ы N2O3.
В качестве соединений, отвечающих основной функции HNO2, можно рассматривать производные нитрозила общей формулы NOX, где Х – одновалентный анион. Хотя галогенидные нитрозилы (Х – F, Cl или Br) и устойчивый лишь ниже –50 °С жёлтый NON3 (т. пл. –57 °С) по свойствам далеки от типичных солей, однако некоторые другие известные вещества того же типа приближаются к ним. Так, кристаллическая структура NOClO4 (легко получаемого действием смеси NO2 + NO на очень крепкую хлорную кислоту) однотипна со структурой NH4ClO4. Соль эта предлагалась в качестве окислителя твёрдого реактивного топлива.
Как соль нитрозила (NOHSO4) следует рассматривать и нитрозилсерную кислоту. Это бесцветное кристаллическое вещество удобно получать действием SO2 на сильно охлаждённую дымящую HNO3. При 73 °С оно плавится и медленно переходит в пиросульфат нитрозила, который может быть получен также прямым взаимодействием NO2 с жидким сернистым газом по схеме:
3 NO2 + 2 SO2 = (NO)2S2O7 + NO.
Интересно, что бесцветный кристаллический (NO)2S2O7 не только плавится, но и кипит без разложения (т. пл. 233, т. кип. 360 °С). Как и другие солеобразные производные нитрозила, он содержит в своём составе ионы NO+. Эти ионы нитрозила (нитрозония) имеют структуру [:NєO:]+ и сильно укороченное расстояние по сравнению с молекулой NO [d(NO) = 106 пм].
Растворы солей нитрозила в растворителях, не взаимодействующих с NO+ (например, NOHSO4 в концентрированной H2SO4), а также некоторые твёрдые его соли (например, NOAlCl4) способны поглощать NO в соответствии с равновесием:
NO++ NO Ы N2O2+.
Образование устойчивого лишь под повышенным давлением NO иона N2O2+ обычно сопровождается появлением синей (реже — фиолетовой) окраски, переходящей при очень сильном охлаждении в красную. Строение этого иона не установлено.
При насыщении NO крепкого раствора К2SO3 образуются бесцветные кристаллы нитрозогидроксиламинсульфоната калия, отвечающего суммарному составу К2SO3·N2O2 и строению ONN(OK)SO3K. В водном растворе соль эта (растворимость 1:10) постепенно разлагается на K2SO4 и N2O.
NO может участвовать в образовании соединений и как отрицательно одновалентный радикал, изоэлектронный возбуждённому состоянию молекулы кислорода. Так, действием NO на раствор Na или Ва в жидком аммиаке были получены NaNO и Ba(NO)2, имеющие солеобразный характер. Аналогичные по составу солеобразные вещества образуются и при взаимодействии NO c амальгамами наиболее активных металлов.
Отвечающее иону NO– водородное соединение (Н–N=O) образуется в результате взаимодействия NO с атомарным водородом (при температуре жидкого воздуха и низком давлении). Получающееся бледно-жёлтое вещество по мере нагревания белеет и выше –95 °С разлагается по схеме:
2 НNO = H2 + 2 NO.
С помощью инфракрасной спектроскопии удалось установить кратковременное существование этого соединения в продуктах фотохимического разложения смесей NO c NH3.
При взаимодействии азотистой кислоты с гидроксиламином по схеме:
HONH2 + ONOH = H2O + HONNOH
образуется азотноватистая кислота (Н2N2O2), которой отвечает структурная формула Н–О–N=N–O–H. Эта кислота представляет собой очень взрывчатое кристаллическое вещество, легкорастворимое в воде, спирте и эфире. При хранении в сухом состоянии и в растворе азотноватистая кислота постепенно распадается по схеме:
Н2N2O2 = H2O + N2O.
Так как распад этот практически необратим, рассматривать N2O в качестве ангидрида азотноватистой кислоты нельзя.
Кислотные свойства Н2N2O2 выражены весьма слабо (К1 = 6·10-8, К2 = 3·10-12), а окислительные у неё практически отсутствуют. Водородом в момент выделения она лишь с трудом восстанавливается до гидразина, под действием кислорода воздуха медленно даёт смесь HNO2 и HNO3, а сильные окислители (например, HMnO4) окисляют её до HNO3. Большинство солей H2N2O2 (называемых азотноватистокислыми или гипонитритами) малорастворимо в воде. Хорошо растворимый Na2N2O2 может быть получен восстановлением NaNO2 с помощью амальгамы натрия. Наиболее характерная для азотноватистой кислоты её малорастворимая (ПР = 1·10-19) жёлтая серебряная соль — Ag2N2O2 (на свету постепенно разлагающаяся). Помимо средних, для H2N2O2 известны и кислые соли. При нагревании и сама кислота, и её соли разлагаются (иногда со взрывом). В частности, Na2N2O2 разлагается при 335 °С с образованием Na2O, NaNO2, NaNO3 и N2.
Довольно сложным путём (взаимодействие CH3ONa, NH2OH и CH3ONO2 в СН3ОН) может быть получен гипонитрат натрия (Na2N2O3) — соль не выделенной в свободном состоянии азотноватой кислоты (H2N2O3).
Взаимодействие растворённого в жидком аммиаке металлического натрия с избытком NaNО2 сопровождается выделением жёлтого порошка:
NaO—N=O NaO—N—ONa
+2 Na ® Ѕ
NaO—N=O NaO—N—ONa
Образующийся продукт является солью гидразотистой кислоты (H4N2O4). Последнюю можно рассматривать как гидразин, в котором все водороды замещены на гидроксильные группы. Подобно H2N2O3, гидроазотистая кислота могла бы считаться гидратом окиси азота (2NO + 2H2O) лишь формально. В свободном состоянии она не получена. Под действием даже следов воды (или при нагревании выше 100 °С) Na4N2O4 разлагается со взрывом.
Окисление гидроксиламиндисульфоната калия [HON(SO2OK)2] в щелочной среде ведёт к образованию фиолетового раствора, из которого могут быть выделены жёлтые кристаллы состава ON(SO2OK)2. Соединение это является сульфопроизводным неизвестной ни в свободном состоянии, ни в виде простых солей гидразотной кислоты (H2NO3 или H4N2O6), которая могла бы формально рассматриваться как гидрат двуокиси азота. Судя по данным магнитных исследований, для кристалла действительна удвоенная формула рассматриваемого соединения, а для раствора — простая. Это указывает на малую прочность связи N—N в вероятной структуре гидроазотной кислоты: (HO)2ON—NO(OH)2.
Основной продукт взаимодействия NО2 с водой — азотная кислота — является одним из важнейших химических соединений. Она потребляется при получении удобрений, органических красителей, пластических масс, взрывчатых веществ и в ряде других производств. Ежегодная мировая выработка азотной кислоты исчисляется миллионами тонн.
Получение азотной кислоты осуществляется в настоящее время каталитическим окислением аммиака. Как было выяснено ещё в 1900 г., при быстром пропускании смеси NН3 с избытком воздуха над нагретым до 800 °С платиновым катализатором по реакции
4 NH3 + 5 O2 = 6 H2O + 4 NO + 900 кДж
образуется окись азота, которая переводится затем в NО2 и НNО3 по приведённым выше реакциям.
Одновременно с приведённой выше реакцией могут протекать различные побочные процессы, в частности,
4 NH3 + 3 O2 = 6 H2O + 2 N2.
Для их предупреждения время контакта газовой смеси с катализатором должно быть очень малым (порядка 1·10-4 с). Катализатор из сплава платины с 5—10 % родия оформляют в виде тонких сеток, сквозь которые и продувается смесь исходных газов. На практике пользуются смесью аммиака с воздухом, содержащей не более 12 объёмн.% NН3. Максимальный выход окиси азота составляет около 98% от теоретического.
Перевод NО в НNО3 представляет значительные технологические трудности, обусловленные главным образом сравнительной медленностью протекания реакции 2 NО + О2 = 2 NО2 и отчасти уменьшением скорости растворения NО2 по мере повышения концентрации НNО3. Для возможно более полного использования NO приходится создавать поглотительные установки большого объёма и с сильно развитой внутренней поверхностью, причём крепость получаемой в обычных условиях HNО3 составляет лишь около 50%. Так как повышение давления ускоряет и окисление NO, и поглощение NО2, необходимый объём поглотительных установок при работе под повышенным давлением резко снижается, а концентрация получаемой HNО3 увеличивается (до 65% при 10 атм). Очень концентрированная (98%) HNО3 может быть получена взаимодействием воды или разбавленной кислоты с жидкой N2О4 и кислородом под давлением 50 атм. Этот “прямой синтез” осуществляют обычно при 70 °С. Получаемую кислоту можно хранить в алюминиевых цистернах. Она используется (как окислитель) в реактивной технике.
С химической стороны интересен впервые осуществлённый в 1901 г. метод получения азотной кислоты “сжиганием воздуха” (т. н. дуговой метод). Более или менее выгодное положение равновесия синтеза NO из элементов достигается лишь при очень высоких температурах и устанавливается при этих условиях практически моментально. В связи с этим задача технического осуществления синтеза NO формулировалась следующим образом: необходимо было изыскать способ нагреть воздух до достаточно высокой температуры и затем очень быстро охладить газовую смесь ниже 1200 °С с тем, чтобы не дать возможности образовавшейся окиси азота распасться обратно на азот и кислород.
При разрешении этой задачи в качестве нагревателя была использована электрическая дуга, дающая температуру около 4000 °С. Если такую дугу поместить между полюсами сильного электромагнита, пламя её образует огненный диск. При быстром пропускании сквозь него струи воздуха последний в момент соприкосновения с пламенем очень сильно нагревается, а затем почти тотчас же охлаждается ниже 1200 °С. В процессе дальнейшего охлаждения газовой смеси NO присоединяет кислород с образованием NO2, из которой затем и может быть получена азотная кислота. На практике образующиеся газы переводили прямо в так называемую норвежскую селитру — Ca(NO3)2, которая затем использовалась в качестве ценного минерального удобрения.
Хотя при техническом проведении процесса выход NO составляет лишь около 2 объёмн.%, это не играет особой роли ввиду отсутствия затрат на исходное сырьё — воздух. Гораздо более существенным недостатком дугового метода является очень большой расход электроэнергии, из-за чего этот метод в настоящее время и не применяется.
Вместе с тем ведутся работы по изучению новых возможностей осуществления “сжигания воздуха” (путём использования регенеративных печей и тепла ядерных реакторов). Если при этом удастся достигнуть достаточно благоприятных техноэкономических показателей, то рассматриваемый метод вновь войдёт в промышленную практику.
“Сжигание воздуха” может служить редким пока примером химического процесса, протекающего в плазме, т. е. газовой фазе, важной составной частью которой являются ионы и электроны. Для земных условий это возникающее за счёт ионизации атомов “четвёртое состояние вещества” не характерно, нов масштабе вселенной оно наиболее распространено (так как из плазмы состоят и звёзды, и межзвёздный газ).
Плазму с температурой не выше десятков тысяч градусов обычно именуют “холодной” (в отличие от “горячей”, отвечающей сотням тысяч и более градусов). Такая холодная плазма, создаваемая в специальных горелках (“плазмотронах”) чаще всего с помощью электрической дуги, перспективна для химии, так как при отвечающих ей температурных условиях — 5000 ч 15000 К — реакции протекают не только очень быстро, но часто в необычных направлениях. Последнее относится прежде всего к сильно эндотермическим процессам (каковым является и синтез NО).
Частичное образование плазмы имеет место при сжигании топлива, и тем относительно в большей степени, чем выше температура горения. Если при этом продукты сгорания охлаждаются достаточно быстро, то они могут содержать NO. Установлено, в частности, что на каждом километре своего пробега легковой автомобиль выделяет с выхлопными газами около 10 г оксида азота(II).
Обычное содержание NO в воздухе у земной поверхности менее 0,005 мг/м3. Благодаря фотохимическим реакциям по суммарным схемам
NO + O2 = NO2 + O и
О + О2 = О3
повышение содержания NO ведёт к накоплению в воздухе NO2 и озона. Эти газы становятся основой “смога” — ядовитого тумана, иногда нависающего в местностях с интенсивным автомобильным движением.
До разработки синтетических методов азотную кислоту получали взаимодействием природной селитры с концентрированной серной кислотой по легко протекающей при нагревании реакции:
NaNO3 + H2SO4 = NaHSO4 + HNO3.
Как видно из уравнения, при этом использовался лишь один водород Н2SO4. Обусловлено это тем, что при высоких температурах, необходимых для введения в реакцию второго водорода, образующаяся HNO3 сильно разлагается, а применяемая для её получения аппаратура быстро изнашивается. Кроме того, вместо легкоплавкого NaHSO4 в этих условиях получается тугоплавкий Na2SO4, удалять который из реакционного пространства весьма трудно.
Безводная азотная кислота представляет собой бесцветную (при хранении быстро желтеющую) жидкость, кипящую при 84 °С. Кипение сопровождается частичным разложением по реакции:
4 HNO3 + 255 кДж = 2 H2O + 4 NO2 + O2.
Растворяясь в перегоняемой кислоте, двуокись азота сообщает ей жёлтую или красную (в зависимости от количества NО2) окраску. Так как NО2 постепенно выделяется из раствора, подобная азотная кислота называется дымящей. Разложение 100%-ной НNО3 медленно идёт на свету уже при обычных температурах.
Молекула азотной кислоты полярна (m = 2,16 ). По данным микроволновой спектроскопии она является плоской. Энергия связи О–N между гидроксилом и нитрогруппой равна 215 кДж/моль. Ион NО3– (в кристаллах NaNO3) представляет собой плоский равносторонний треугольник с азотом в центре [d(NO) = 122 пм].
В безводной азотной кислоте имеют место следующие равновесия:
3 HNO3Ы H3O+ + NO3– + N2O5Ы H3O+ + 2 NO– + NO2+.
По мере разбавления водой равновесия эти смещаются влево и уступают место нормальной ионизации:
H2O + HNO3Ы H3O+ + NO3-.
Однако даже обычная концентрированная НNО3 содержит, по-видимому, небольшие количества и N2О5, и катиона нитронила (нитрония) NО2+. Последний имеет линейную структуру [O=N=O]+ с ядерным расстоянием d(NO) = 115 пм. Для него вероятно следующее распределение электронной плотности: dN = +0,60, dO = +0,20.
Безводная НNО3 очень хорошо растворяет жидкую N2О4 и сама несколько растворима в ней. Насыщенная обоими компонентами система HNO3 — N2О4 распадается на два жидких слоя, из которых при 20 °С один содержит 44% HNО3 и 56% N2О4, а другой — 93% N2О4 и 7% НNО3.
Растворение N2О4 в безводной HNО3 ведёт к повышению плотности, понижению температуры замерзания (18%-ный раствор замерзает лишь при –73 °С) и резкому усилению окислительной активности. Такие растворы используются в реактивной технике. Растворы некоторых органических веществ (например, нитробензола) в безводной HNО3 применяются как жидкие взрывчатые вещества.
Безводная HNО3 является хорошим растворителем для некоторых солей (главным образом нитратов одновалентных металлов) и свободных кислот. Подобные растворы обладают высокой электропроводностью, что указывает на наличие ионизации, например, по схеме:
KNO3 + HNO3Ы K+ + [H(NO3)2]–.
Комплексный анион [H(NO3)2]– по строению аналогичен иону HF2– и имеет плоскую структуру с расстоянием d(ОО) = 245 пм в группировке О···Н+···О. Отвечающие структурному типу М[H(NO3)2] двойные нитраты Cs, Rb, K (а также некоторых комплексных катионов) были выделены в твёрдом состоянии.
С водой HNО3 смешивается в любых соотношениях. Применяемая в лабораторной практике реактивная азотная кислота содержит около 65% HNО3 и имеет плотность 1,40 г/см3. По составу она приблизительно соответствует формуле HNO3·2H2O.
Снежно-белые кристаллы безводной азотной кислоты имеют плотность 1,52 г/см3 и плавятся при –41 °С. Известны два кристаллогидрата: HNO3·H2O и HNO3·3H2O. Максимальной электропроводностью обладает 30%-ный раствор HNО3. Смешиванием предварительно охлаждённой до 0 °С концентрированной HNО3 со снегом (1:2 по массе) может быть достигнуто охлаждение до –56 °С.
Ниже приводятся данные, иллюстрирующие зависимость плотности и температуры кипения водных растворов от процентного содержания в них HNО3:
HNО3, % ············· 100 94,1 86,0 68,4 65,3 47,5 24,8
Плотность, г/см3········ 1,51 1,49 1,47 1,41 1,40 1,30 1,15
Температура кипения, °С · 86 99 115 122 119 113 104
Из этих данных видно, что максимальную температуру кипения (122 °С) имеет раствор, содержащий 68,4% HNО3. Такой раствор будет в конце концов получаться при упаривании как более разбавленной, так и более концентрированной кислоты. Для получения последней удобно пользоваться повторной перегонкой обычной 65%-ной HNО3 из смеси с концентрированной H2SO4.
Формы азотной кислоты с повышенным содержанием химически связанной воды — NO(OH)3 и N(OH)5 — в индивидуальном состоянии неизвестны. Отвечающие им по составу соли натрия (Na2HNO4, Na3NO4, Na3H2NO5) образуются при сплавлении NaNO3 c Na2O или NaOH. Однако свойства получаемых веществ говорят в пользу того, что они представляют собой не индивидуальные соединения, а простые сплавы. То же относится и к продукту протекающей при 250 °С в вакууме реакции
10 Na + 8 NaNO3 = N2 + 6 Na3NO4.
С химической стороны концентрированная азотная кислота характеризуется прежде всего сильно выраженными окислительными свойствами. При этом основным конечным продуктом восстановления не очень крепкой HNО3 является NO, а концентрированной — NО2.
Все часто встречающиеся в практике металлы, за исключением Au и Pt, переводятся концентрированной азотной кислотой в оксиды. Если последние растворимы в HNO3, то образуются азотнокислые соли. По этой схеме азотная кислота растворяет и такие стоящие в ряду напряжений правее водорода металлы, как Сu, Hg и Ag.
Некоторые металлы, бурно реагирующие с разбавленной азотной кислотой, практически не взаимодействуют с концентрированной (и особенно дымящей). Обусловлено это тем, что на их поверхности образуется очень тонкий, но плотный слой нерастворимого в концентрированной кислоте оксида, защищающего металл от дальнейшего разъедания. Такая “пассивность” особенно важна в случае Fe, так как позволяет перевозить концентрированную HNО3 в стальных цистернах.
Весьма энергично действует концентрированная (особенно дымящая) азотная кислота на некоторые неметаллы. Так, сера окисляется ею при кипячении до H2SO4, уголь — до СО2 и т. д. Животные и растительные ткани при действии HNО3 разрушаются.
Основным первоначальным продуктом восстановления концентрированной HNО3 является, по-видимому, азотистая кислота. Если процесс проводится в не очень крепких растворах, то из образующихся при её распаде газов выделяется только NO (так как NO2, реагируя с водой, даёт HNО3 и NО). Однако по мере повышения концентрации всё большее значение начинает приобретать обратимость реакции
3 NO2 + H2O Ы 2 HNO3 + NO + 70 кДж.
При эквивалентных соотношениях реагирующих веществ равновесие её смещено вправо, но последовательное повышение концентрации HNО3 всё более смещает его влево. Поэтому основным конечным продуктом восстановления концентрированной HNО3 и становится NO2, а не NO. Само собой разумеется, что при этом может частично образовываться и N2О3.
Реакции окисления азотной кислотой являются аутокаталитическими процессами, причём роль катализатора играет диоксид азота. Значительно более сильное окислительное действие дымящей HNО3 по сравнению с обычной обусловлено именно наличием в первой больших количеств NО2. Ход реакции может быть выражен следующими элементарными процессами:
NO2 + e ® NO2– H+ + NO2–Ы HNO2
HNO3 + HNO2Ы H2O + 2 NO2.
Для первоначального введения в азотную кислоту её окислительного катализатора можно воспользоваться кристаллом какого-нибудь нитрита.
Напротив, освободить HNО3 от растворённых оксидов азота можно добавлением небольшого количества сульфаминовой кислоты. Основная реакция идёт по уравнению:
HNO2 + NH2SO2OH = N2 + H2SO4 + H2O.
Обработанная таким образом азотная кислота в разбавленном растворе не окисляет йодистый водород.
Для отличия азотной кислоты от азотистой важно их отношение к HI. В то время как азотистая кислота тотчас окисляет йодистый водород до свободного иода, разбавленная азотная кислота на HI не действует. Напротив, концентрированная азотная кислота окисляет не только HI, но и HCl. Однако в последнем случае реакция обратима:
HNO3 + 3 HCl Ы 2 H2O + NOCl + Cl2.
Смесь концентрированной HNO3 c концентрированной HCl называют обычно “царской водкой”. Она действует значительно энергичнее, чем каждая из этих кислот в отдельности. Так, даже Au и Pt легко растворяются в царской водке с образованием соответствующих хлористых соединений по схемам:
Au + HNO3 + 3 HCl = AuCl3 + NO + 2 H2O
3 Pt + 4 HNO3 + 12 HCl = 3 PtCl4 + 4 NO + 8 H2O.
Активным действующим началом царской водки является хлор в момент выделения.
Приведённые в основном тексте реакции растворения Au и Pt в царской водке передают лишь основной процесс. На последний налагается образование комплексных кислот и их нитрозосолей по схемам:
HCI + AuCl3 = H[AuCl4] и NOCl + AuCl3 = NO[AuCl4],
2 HCl + PtCl4 = H2[PtCl6] и 2 NOCl + PtCl4 = (NO)2[PtCl6].
Относительное значение этих вторичных реакций зависит от соотношения концентраций соляной и азотной кислот.
Азотная кислота является не только сильным окислителем, но и сильной кислотой. При последовательном разбавлении раствора окислительные свойства быстро ослабляется, а кислотные усиливается, поэтому реакции многих металлов с разбавленной HNО3 протекают по общему типу, т. е. с вытеснением водорода. Однако последний обычно не выделяется, а расходуется на восстановление избытка HNО3 до производных более низкой значности азота, вплоть до NН3. Как правило, получается смесь различных продуктов восстановления.
В достаточно концентрированных растворах азотной кислоты (выше 2 М) с помощью оптических методов можно непосредственно установить существование её недиссоциированных молекул. При этих условиях удаётся также оценить константу кислотной диссоциации HNО3, причём полученное значение К = 20. Недавно было показано, что диссоциирующей фактически является орто-форма азотной кислоты — Н3NО4 (отвечающая тетраэдрической координации атома азота).
Характер продуктов восстановления HNО3 сильно зависит от ряда факторов — концентрации кислоты, природы восстановителя, температуры и т. д. Чем левее в ряду напряжений располагается металл (и разбавленнее кислота), тем больше относительное содержание аммонийных солей в продуктах реакции. Кипячением в щелочной среде с порошком алюминия нитраты могут быть количественно восстановлены до аммиака. Реакция идёт по уравнению:
8 Al + 3 NaNO3 + 5 NaOH + 2 H2O = 8 NaAlO2 + 3 NH3.
Как очень сильная одноосновная кислота HNО3 образует вполне устойчивые соли. Подобно самому иону NO3-, большинство нитратов бесцветно. Почти все азотнокислые соли хорошо растворимы в воде. Многие из них находят разнообразное практическое применение.
Проявление оснувной функции HNО3 имеет место при её взаимодействии с HF и некоторыми другими кислотами. Во всех подобных случаях диссоциация HNО3 протекает с образованием катиона нитронила по схемам, например:
HNO3 + 2 HClO4Ы H3O+ + NO2+ + 2 ClO4-или
HNO3 + 2 H2SO4Ы H3O+ + NO2+ + 2 HSO4-.
Некоторые соли нитронила (NO2ClO4, NO2HSO4 и др.) были получены в твёрдом состоянии. Водой они тотчас гидролизуются до соответствующих свободных кислот. Перхлорат нитронила (NO2ClO4) был предложен в качестве окислителя твёрдого реактивного топлива.
При взаимодействии по уравнению
2 NO2 + F2 = 2 NO2F
образуется газообразный (т. пл. –166 °С, т. кип. –72 °С) фтористый нитронил (нитрил). Он может быть получен также по реакции:
F2 + NaNO2 = NaF + FNO2.
Молекула FNО2 полярна и имеет структуру плоского треугольника [d(FN) = 147, d(NO) = 118 пм, РONO = 136°]. Энергии связей оцениваются в 190 (FN) и 255 кДж/моль (NO). Фтористый нитронил не действует на водород, серу и уголь, но, подобно NOF, переводит кремний, бор, фосфор и многие металлы в их фтористые соединения. Водой он разлагается на HF и азотную кислоту. С фторидами некоторых других элементов NO2F образует комплексы, примерами которых могут служить бесцветные NO2MoF7 и NO2WF7.
Взаимодействием N2О4 с F2 при –30 °С в алюминиевом реакторе был синтезирован изомерный нитронилфториду нитрозилгипофторит — ONOF. Он представляет собой бесцветный газ, при хранении более или менее быстро переходящий в нитронилфторид.
Аналогичное NO2F хлористое производное (NO2Cl) может быть получено действием озона на NOCl или по реакции:
HSO3Cl + HNO3 = H2SO4 + ClNO2.
Его плоская молекула характеризуется структурными параметрами d(ClN) = 184, d(NO) = 120 пм, РONO = 130° и малой полярностью. Нитронилхлорид представляет собой бесцветный газ (т. пл. –145, т. кип. –14 °С). при хранении он медленно распадается на NO2 и Cl2, раствором щёлочи разлагается по схеме
NO2Cl + 2 NaOH = NaNO2 + NaOCl + H2O,
а с водой даёт HNO3 и HСl (видимо, в результате вторичной реакцииHNO2 и HOCl). Бромистый нитронил не получен. По-видимому, при обычной температуре он может существовать только в газовой фазе, содержащей одновременно избыток и NO2, и Br2. Фторсульфонат нитронила интересен как соль, дающая при гидролизе сразу три кислоты:
NO2SO3F + 2 H2O = HNO3 + H2SO4 + HF.
Соответствующее нитрилам амидное производное — нитрамид (NH2NO2) является, по-видимому, промежуточным продуктом при термическом распаде нитрата аммония:
NH4NO3® H2O + NH2NO2® 2 H2O + N2O.
В свободном состоянии он может быть получен лишь сложным косвенным путём. Молекула его имеет большой дипольный момент и следующее строение: d(HN) = 101, d(NN) = 143, d(NO) = 121 пм, Р(ONO) = 130° с углом 52° между плоскостями H2N и NО2.
Нитрамид представляет собой малоустойчивое бесцветное кристаллическое вещество (т. пл. 75 °С с разл.), легкорастворимое в воде, спирте и эфире. В водном растворе нитрамид имеет слабо выраженный кислотный характер (К = 3·10–7). Однако взаимодействие его со щелочами ведёт не к образованию солей, а к распаду нитрамида на H2O и N2О. Выделена была только одна его соль — HgNNO2.
Действием электроразряда на смесь NF3 и О2 может быть получен оксонитротрифторид — ONF3. Почти неполярная молекула этого бесцветного газа (т. пл. –160, т. кип. –85 °С) имеет строение несколько искажённого тетраэдра с атомом азота около центра. Для него принята формула O=NF3 c пятиковалентным азотом.
При обычных условиях ONF3 устойчив (в сосудах из никеля — даже до 300 °С) и практически не реагирует ни со стеклом, ни с водой. Однако он является сильным окислителем, проявляющим преимущественно фторирующее действие (с восстановлением до ONF). Например, Cl2 окисляется им до ClF, а N2O4 переводится в NO2F. Вместе с тем он способен образовывать комплексные соединения с SbF5 или AsF5 (но не PF5) типа [ONF2][ЭF6], в которых катион ONF2+ имеет форму плоского треугольника. Этот катион гораздо более реакционноспособен, чем ONF3 (где атом азота практически изолирован от внешних воздействий).
Взаимодействием безводной HNО3 с фтором может быть получен продукт замещения на фтор водорода азотной кислоты — фторнитрат. Его образование идёт по уравнению:
F2 + HNO3 = HF + FNO3.
Соединение это можно рассматривать и как гипофторит нитронила — NO2OF [d(OF) = 142 пм].
Фторнитрат представляет собой бесцветный газ с характерным удушливым запахом (т. пл. –175, т. кип. –46 °С). При хранении он постепенно разлагается, а при соприкосновении его с некоторыми органическими веществами (спирт, эфир и т. п.) происходит взрыв. В воде фторнитрат довольно хорошо растворим, причём гидролизуется сравнительно медленно. При взаимодействии его с разбавленным раствором NaOH реакция идёт, по-видимому, по уравнению:
2 FNO3 + 2 NaOH = 2 NaNO3 + F2O + H2O.
Взаимодействие с концентрированным раствором NaOH сопровождается выделением кислорода. С уксусной кислотой, перманганатом и большинством металлов FNO3 не взаимодействует, а с SbCl5 и TiCl3 даёт жёлтые твёрдые продукты присоединения.
Аналогичный фторнитрату бесцветный ClNO3 (т. пл. –107, т. кип. +18 °С) может быть получен по реакции:
2 Cl2O + 2 NO2 = 2 ClNO3 + Cl2.
Взаимодействие его со щёлочью сопровождается образованием соответствующих нитрата и гипохлорита. Были получены также аналогичные производные брома и иода.
При достаточном нагревании нитратов они разлагаются, причём характер распада зависит от природы катиона. Соли наиболее активных металлов (расположенных в ряду напряжений левее Mg) с отщеплением кислорода переходят в соответствующие нитриты, соли менее активных (Mg — Cu) распадаются с образованием оксидов, и ещё менее активных (правее Cu) — с образованием свободных металлов. Примерами могут служить реакции:
2 NaNO3 = 2 NaNO2 + O2,
2 Pb(NO3)2 = 2 PbO + 4 NO2 + O2,
2 AgNO3 = 2 Ag + 2 NO2 + O2.
Неодинаковый характер протекания этих реакций обусловлен различной устойчивостью соответствующих нитритов и оксидов при температурах распада: в этих условиях для Na ещё устойчив нитрит, для Pb он уже неустойчив, но ещё устойчив оксид, а для Ag неустойчиво и то и другое соединение.
Ввиду лёгкости отдачи кислорода солями азотной кислоты при высоких температурах, смеси их с горючими веществами сгорают чрезвычайно быстро. На этом основано применение нитратов в пиротехнике и для изготовления чёрного пороха.
Чёрный порох представляет собой тесную смесь KNO3 с серой и углём. Смеси такого типа были, по-видимому, изобретены около 100 г. до н. э. в Китае. Их взрывное действие трактовалось как результат внезапного соединения противоположных начал “ян” и “инь”, причём носителем “ян” считалась сера, а носителем “инь” — селитра. В Европе порох стал известен около 1200 г. Первый точный рецепт его изготовления даётся в составленной до 1250 г. “Книге огня” Марка Грека следующим образом: “Возьми 1 фунт живой серы, 2 фунта липового или ивового угля, 6 фунтов селитры. Очень мелко разотри эти три вещества на мраморной доске и смешай”.
Так называемый “нормальный порох” (68 KNO3, 15 S и 17% С) приблизительно отвечает составу 2 KNO3 + 3 C + S, вообще же относительные количества составных частей в отдельных сортах колеблются. Реакция горения чёрного пороха протекает по суммарному уравнению:
2 KNO3 + 3 C + S = N2 + 3 CO2 + K2S + 615 кДж,
хотя частично образуются также CO, K2CO3, K2SO4 и K2S2. Ввиду наличия среди продуктов сгорания твёрдых веществ, взрыв чёрного пороха сопровождается выделением дыма. Напротив, пироксилин твёрдых продуктов сгорания не образует, а потому приготовленные на его основе пороха сгорают без дыма (“бездымные пороха”).
Большинство взрывчатых веществ готовят взаимодействием при определённых условиях некоторых органических соединений с азотной кислотой. Например, исходя из целлюлозы (обычно в виде хлопка), получают пироксилин по реакции:
C6H10O5 + 3 HNO3 = 3 H2O + C6H7O2(ONO2)3.
Он способен разлагаться по схеме:
2 C6H7O2(ONO2)3 = 3 N2 + 9 CO + 3 CO2 + 7 H2O.
Так как этот распад протекает экзотермически, вода выделяется также в газообразном состоянии. Мгновенное образование из небольшого объёма твёрдого пироксилина громадного объёма газообразных веществ и обусловливает взрывной эффект. В частности, каждый килограмм пироксилина может совершить при взрыве работу, равную 470 тыс. кГм.
По характеру действия взрывчатые вещества делятся на инициирующие, бризантные и метательные. Первые (детонаторы типа азида свинца) характеризуются наибольшей скоростью разложения, которое может быть вызвано механическим воздействием — ударом, наколом и т. п. Инициирующие взрывчатые вещества применяются для снаряжения взрывателей.
Бризантные (иначе дробящие) взрывчатые вещества характеризуются меньшей скоростью разложения, которая всё же очень велика. Например, скорость распространения взрыва пироксилина составляет 6300 м/с. При таком почти мгновенном разложении взрывчатого вещества образуется громадный объём газов, которые и оказывают резкое давление на окружающую среду. Бризантные взрывчатые вещества применяются для снаряжения снарядов, мин, авиабомб и т. д., а также при различных подрывных работах. Обычно они взрываются только от детонации, т. е. от происходящего в непосредственной близости взрыва инициирующего вещества.
Метательные взрывчатые вещества взрываются только от детонации и характеризуются сравнительной медленностью своего разложения. Например, скорость распространения взрыва чёрного пороха составляет всего 300—400 м/с. Подобные взрывчатые вещества применяются для снаряжения ружейных и орудийных зарядов. Вследствие сравнительно малой скорости разложения метательного взрывчатого вещества, пуля или снаряд за время взрыва успевает покинуть ствол и открыть вход образующимся газам. Напротив, при снаряжении патрона пироксилином ствол в момент выстрела был бы разорван. поэтому изготовление бездымных порохов на базе пироксилина и сводится главным образом к уменьшению скорости его разложения путём добавки к нему веществ, не имеющих взрывчатого характера.
Возникающие при взрывах ударные волны находят ряд не существовавших ранее технических применений. Например, ими уже довольно широко пользуются для штамповки стальных деталей. Интересны исследования по созданию новых источников света, действующих лишь миллионные доли секунды, но обладающих громадной яркостью. Принцип их получения прост: небольшая ёмкость, содержащая газ (например, аргон) под обычным давлением, отделяется тонкой плёнкой от заряда взрывчатого вещества. Направленный взрыве которого, в газовой среде за счёт резкого сжатия газа создаёт высокотемпературную плазму. Этим методом удавалось получить нагретую до 90000 К плотную плазму, по яркости (на единицу поверхности) в 50 тыс. раз превосходящую Солнце.
Отвечающий азотной кислоте ангидрид может быть получен взаимодействием NO2 с озоном:
2 NO2 + O3 = O2 + N2O5 + 250 кДж.
Азотный ангидрид (N2O5) представляет собой бесцветные, очень летучие кристаллы. Последние образованы ионами NO2+ и NO3–, а в парах ангидрид состоит из отдельных молекул, строение которых отвечает формуле O2N–O–NO2. Он крайне неустойчив и уже при обычных условиях медленно разлагается на двуокись азота и кислород. Будучи сильным окислителем, азотный ангидрид бурно реагирует со способными окисляться веществами. С водой он образует азотную кислоту.
Азотный ангидрид может быть получен дегидратацией HNО3 посредством P2O5 или пропусканием сухого хлора над сухим AgNO3. Последняя реакция протекает по уравнению:
2 Cl2 + 4 AgNO3 = 4 AgCl + 2 N2O5 + O2.
МолекулаN2O5 полярна (m = 1,39 для раствора в CCl4) и слагается из двух групп NO2 [d(NO) = 121 пм, РONO = 134°], связанных друг с другом атомом кислорода [d(ON) = 146 пм при угле 95° между плоскостями групп NO2]. Давление пара азотного ангидрида (т. возг. 32 °С) составляет при обычных условиях около 300 мм рт. ст. В запаянной трубке он плавится при 41 °С.
Теплота образования кристаллического N2О5 из элементов равна 40 кДж/моль, а теплота его возгонки 55 кДж/моль. Результаты изучения скорости разложения паров азотного ангидрида по суммарному уравнению
2 N2O5 = O2 + 2 N2O4
указывают на то, что реакция является не бимолекулярной, а мономолекулярной. Обусловлено это тем, что общая скорость химического превращения определяется его наиболее медленной стадией. В действительности, при распаде N2O5, по-видимому, имеют место следующие элементарные процессы:
N2O5Ы NO2 + NO3 (быстрая реакция),
NO3® NO + O2 (медленная),
NO3 + NO ® N2O4 (быстрая).
Энергия активации разложения N2О5 составляет 100 кДж/моль. При 0 °С половина его исходного количества распадается за 10 дней, а при 20 °С — за 10 часов.
Взаимодействием N2О5 со 100%-ной Н2О2 при –80 °С может быть получено очень взрывчатое вещество с запахом хлорной извести. Этому малоизученному соединению приписывают формулу надазотной кислоты — HNО4. В растворе она частично образуется при взаимодействии 100%-ной Н2О2 с обычной концентрированной HNО3 по обратимой реакции:
Н2О2 + HNO3Ы H2O + HNO4.
Применение 70%-ной и ещё более крепкой HNО3 вызывает разложение (сопровождающееся взрывом), а при концентрациях HNО3 ниже 20% происходит полный гидролиз HOONO2.
При испарении смеси N2О4 с избытком жидкого озона получается очень неустойчивое белое вещество, отвечающее по составу простейшей формуле NO3. Так как взаимодействие этой перекиси азота с водой медленно идёт по схеме
2 NO3 + H2O = HNO3 + HNO2 + O2,
ей соответствует, вероятно, формула O2N–O–O–NO2. Следует отметить, что имеющиеся данные по перекисным производным азота довольно противоречивы.
Круговорот азота.
Азот исключительно важен для жизни, так как основа живых организмов — белки — содержат его около 17%. В природе этот элемент непрерывно проходит определённый цикл превращений.
Основной первичной формой существования азота на земной поверхности был, по-видимому, аммиак, выделявшийся из горячих земных недр. Впоследствии он дал начало свободному азоту атмосферы, частично за счёт разложения на элементы под действием ультрафиолетового излучения Солнца, частично за счёт окисления. Следы аммиака (примерно 2·10-6 объёмн.%) постоянно содержатся в современной нам атмосфере.
Частые и мощные электрические разряды в тёплой и очень влажной атмосфере отдалённых геологических эпох обусловливали частичное связывание атмосферного азота в NO. Монооксид азота превращается затем в NO2 и азотную кислоту, которая вместе с дождём выпадала на Землю и нейтрализовалась солями более слабых кислот (например, углекислыми). Таким образом, первичным образованием кислородных соединений азота Земля обязана грозам.
С развитием органической жизни как аммиак, так и соли азотной кислоты стали служить материалом для выработки растениями белковых веществ. Растения частично поедаются травоядными животными, а последние служат пищей плотоядным животным. Экскременты тех и других, их трупы, а также останки самих растений возвращают почве взятый из неё связанный азот. Под воздействием особых видов бактерий эти останки “гниют”, т.е. претерпевают ряд сложных биохимических изменений, причём в конечном счёте их азот переходит в аммиак и соли аммония.
Конечные продукты гниения частично вновь усваиваются растениями, частично подвергаются в почве дальнейшему превращению в соли азотной кислоты. Обусловливающий этот переход природный процесс носит общее название “нитрификации” и протекает под воздействием двух видов микроорганизмов: нитрозобактерий и нитробактерий.
И для тех, и для других окисление аммиака и солей аммония за счёт кислорода воздуха служит источником необходимой им для жизни энергии. При этом между обоими видами бактерий существует строгое “разделение труда”. Первые вызывают окисление аммиака только до азотистой кислоты по схеме:
2 NH3 + 3 O2 = 2 HNO2 + 2 H2O + 715 кДж
вторые — окисление азотистой кислоты до азотной:
2 HNO2 + O2 = 2 HNO3 + 176 кДж.
Образующаяся азотная кислота переводит углекислые соли почвы в нитраты, которые затем вновь усваиваются растениями и т.д. Таким образом основной цикл превращений связанного азота замыкается.
В этом основном цикле имеются, однако, серьёзные источники потерь связанного азота. Некоторая его часть всегда выделяется в свободном состоянии и при гниении, и при нитрификации. Подобным же образом связанный в виде органических соединений азот переходит в свободный при лесных и степных пожарах.
Другой источник потерь связан с жизнедеятельностью “денитрифицирующих” бактерий, получающих необходимую им для жизни энергию за счёт окисления органических веществ кислородом по суммарной схеме (С — углерод органических веществ):
5 С + 4 КNO3 = 2 K2CO3 + 3 CO2 + 2 N2 + 1500 кДж.
Деятельность этих бактерий ведёт, таким образом, к непосредственному переводу нитратов в свободный азот, который выходит из круговорота.
Наряду с источниками потерь в природе имеются и источники пополнения. В этом направлении и теперь продолжают действовать атмосферные электрические разряды. Установлено, что на земном шаре ежесекундно разряжаются около 2000 молний. Приблизительно подсчитано, что таким путём в почву ежегодно вносится до 15 кг связанного азота на гектар.
Другим источником пополнения является жизнедеятельность “азотобактерий”, способных в присутствии органических веществ переводить свободный азот в аммиак. Вызванный ими процесс протекает по суммарной схеме (С — углерод органических веществ):
2 N2 + 6 H2O + 3 C + 351 кДж = 3 СО2 + 4 NH3.
При благоприятных условиях азотобактериии способны за год накопить в почве до 30 кг связанного азота на гектар.
Ещё гораздо большие количества свободного азота могут связать “клубневые” бактерии, колонии которых образуют характерные наросты на корнях растений семейства бобовых: клевера, люцерны, люпина, гороха, фасоли и др. Питаясь соками растения, они одновременно переводят свободный азот атмосферы в азотные соединения, которые усваиваются растением-хозяином. Это позволяет растениям семейства бобовых успешно развиваться на почвах, бедных соединениями связанного азота. Наиболее благоприятны для развития клубеньковых бактерий почвы с рН = 6ч7.
Культивация бобовых растений является мощным средством общего поднятия урожайности, так как накапливаемый их корнями азот сохраняется в почве. Так, клевер или люпин даёт примерно 150 кг связанного азота на 1 га. “Каждый куст люпина (или другого бобового) есть в сущности миниатюрный завод по утилизации атмосферного азота, работающий даром за счёт солнечной энергии” (Д.Н. Прянишников). С химической стороны процесс фиксации азота клубеньковыми бактериями ещё недостаточно выяснен, но ведёт к образованию аммиака.
Громадную роль в круговороте азота играет жизнедеятельность бактериального мира (каждый грамм почвы содержит миллиарды микроорганизмов). Это обстоятельство почти исключает возможность количественного подсчёта круговорота. Всё же можно думать, что в условиях свободного протекания природных процессов поддерживается некоторое равновесие и общее количество соединений связанного азота меняется во времени лишь незначительно.
Существенные осложнения вносит в круговорот азота сознательная деятельность человека. На протяжении длительных эпох она сводилась исключительно к переводу азотных соединений в свободный азот атмосферы, т. е. к выводу его из круговорота.
Главнейшую роль при этом играл огонь. При горении топлива все содержащиеся в нём азотные соединения разлагаются с выделением свободного азота. Помимо поддержания огня у своего очага, человек с целью расчистки места для посевов часто выжигал целые леса и степи. Впоследствии к этому присоединилось колоссальное потребление топлива для промышленных целей. В общем, за время своего существования человечество сжиганием топлива вывело из круговорота громадные количества связанного азота.
Уже в новейшую эпоху заметное значение приобрёл другой источник его потерь — применение взрывчатых веществ. Последние во многих случаях (горные разработки и т.д.) значительно облегчают механическую работу человека и поэтому используются в большом масштабе. Между тем взрывчатые вещества обычно содержат кислородные соединения азота, причём последний при взрыве выделяется в свободном состоянии и выходит из круговорота.
Обратное направление сознательной деятельности человека — введение в круговорот свободного азота атмосферы — могло идти двумя путями: использованием жизнедеятельности бактерий и искусственным связыванием азота атмосферы. Первый из них — введение в севооборот бобовых растений — был известен ещё древним грекам, римлянам и китайцам. Важную роль играет он и в правильно поставленном сельском хозяйстве настоящего времени.
Второй путь — искусственное связывание азота атмосферы — получил своё развитие только в текущем столетии. Сюда относятся методы “сжигания воздуха”, синтеза аммиака и цианамидный. Хотя все они могут рассматриваться как большое достижение химической техники, однако сопоставление с работой бактерий наглядно вскрывает грубость и несовершенство этих методов. Действительно, относительные затраты энергии для получения одинакового количества связанного азота по каждому из них составляют:
“сжигание воздуха” цианамидный синтез аммиака
9 2 1
Синтез аммиака является, следовательно, самым совершенным из всех трёх методов. Но, как известно, он идёт лишь при высоких температурах (что резко сказывается на уменьшении выхода аммиака) и больших давлениях. Между тем бактерии проводят тот же синтез при обычных условиях. Из этого следует, что в методе синтеза аммиака скрыты большие возможности его дальнейшего совершенствования.
Человек пока не научился искусственно осуществлять только те направления в круговороте азота, которые связаны с синтезом сложных белковых веществ (хотя на пути к этому выполнены уже многие подготовительные работы).
До сих пор рассматривался общий баланс азота в природе. Между тем практически более важен баланс азотных соединений на тех отдельных участках земной поверхности, которые используются в качестве полей для посевов. С этой точки зрения громадное значение имеют те направления человеческой деятельности, которые, не изменяя общего баланса, ведут к перемещению связанного азота (а также двух других важнейших для жизни растений элементов — фосфора и калия).
Культурные растения извлекают из почвы следующие средние количества связанного азота (в кг на тонну):
Озимая рожь Яровая пшеница Картофель Сахарная свёкла
зерно солома зерно солома клубни ботва корни ботва
14 4,5 20,5 6 3 3 2 3
Лишь малая часть урожая сельскохозяйственных культур в той или иной форме потребляется на местах произрастания. Большая часть используется для питания населения и для промышленной переработки. При этом азот и другие извлекаемые растениями элементы уже не попадают обратно на поля. В результате почва постепенно истощается и урожаи начинают из года в год падать. Для предотвращения этого необходимо внесение на поля достаточного количества нужных растениям элементов (главным образом N, P и К) в форме минеральных удобрений.
33
Водород.
Водород — наиболее лёгкий из всех элементов. Он стоит в начале периодической системы и не может быть отнесён к какой-либо определённой её группе. Его особое положение в системе вызвано тем, что первый период содержит только два элемента — водород и гелий, а не так как остальные периоды — 8 и больше элементов. Водород объединяет признаки первой и предпоследней (VII) групп. Но при этом существуют и большие различия в его отношении к щелочным металлам и галогенам. Химические свойства, которыми он напоминает щелочные металлы (за исключением его валентности), обусловлены совсем другими обстоятельствами, чем у щелочных металлов. Напротив, свойства, которые определяют его сродство к галогенам, у водорода объясняются теми же, что и у последних, причинами. Поэтому водород — это галоген, который вследствие своего особого положения в качестве первого члена периодической системы проявляет в химическом отношении некоторое внешнее сходство со щелочными металлами.
Как и галогены, он является неметаллом: в элементарном состоянии образует двухатомные молекулы, в которых атомы водорода (как и галогенов) связаны простой связью. Энергия диссоциации молекул постепенно убывает в ряду Н-Сl-Br-F-I. Так же как и галогены, водород может вступать в качестве электроположительного иона, т. е. он обладает сродством к электрону (при присоединении электрона к нейтральному атому выделяется энергия). Так же как и водород, галогены в соединениях, где они отрицательно заряжены, исключительно одновалентны. Соединения водорода с металлами, в которых водород является электроотрицательной составной частью по строению и характеру связи (ионные гидриды) аналогичны галогенидам.
Уже одно то обстоятельство, что водород в качестве первого члена в ряду элементов может терять только один электрон, доказывает, что он по многим свойствам сильно отличается от галогенов. В случае отщепления от атома водорода электрона остаётся очень маленькое ядро атома — протон. Это отличает водород от всех остальных элементов периодической системы. Для всех остальных элементов диаметр ионов, полученных после отщепления валентных электронов, лежит в пределах 20-330 пм; величина же водородного ядра составляет лишь 10-3 пм. При соединении такого исключительно малого ядра с отрицательно заряженным ионом выделяется особенно много энергии. Это относится и к образованию иона гидроксония Н3О+ в водных растворах, где ионы водорода соединяются с молекулами воды, и к другим аналогичным реакциям. Поэтому водород, несмотря на высокий ионизационный потенциал, ведёт себя как довольно сильно электроположительный элемент. Водород проявляет склонность к образованию малополярных ковалентных связей и, в соответствии с этим, легколетучих соединений. Вследствие летучести этих соединений равновесия реакций с их участием смещены в сторону их образования. На этом основана большая восстановительная способность водорода.
Щелочные металлы, как и водород, могут быть только одновалентными, но, в отличие от него, исключительно электроположительными, при этом водород не является металлом и обладает, в отличие от щелочных металлов, способностью заряжаться отрицательно. Щелочные металлы имеют самые низкие из всех элементов потенциалы ионизации и находятся левее всех в ряду напряжений, водород же в этом ряду находится значительно правее и имеет даже более высокий потенциал ионизации, чем благородные металлы.
Водород является одним из наиболее распространённых элементов — его доля составляет 0,88% от массы всех трёх оболочек земной коры (атмосферы, гидросферы и литосферы), что при пересчёте на атомные проценты даёт цифру 15,5.
Основное количество этого элемента находится в связанном состоянии. Так, вода содержит его около 11 вес. %, глина — около 1,5% и т. д. В виде соединений с углеродом водород входит в состав нефти, горючих природных газов и всех организмов.
Свободный водород состоит из молекул Н2. Он часто содержится в вулканических газах. Частично он образуется также при разложении некоторых органических остатков. Небольшие его количества выделяются зелёными растениями. Атмосфера содержит около 5•10-5 объёмн. % водорода.
Водород был впервые описан в 1766 г. Кавендишем, который получал его действием железа и некоторых других металлов на разбавленную серную или соляную кислоту (сама эта реакция была известна значительно раньше). Полученный лёгкий газ Кавендиш принял сперва за флогистон, а затем (1781 г.) за соединение флогистона с водой. Разложение воды раскалённым железом было впервые (1783) проведено Лавуазье (при продувании струи пара через нагретый до красного каления оружейный ствол). Современное название дал этому элементу Лавуазье (1783 г.). Разложение воды электрическим током было осуществлено в 1789 г.
Водород состоит из смеси изотопов с массовыми числами 1 и 2 (1Н и 2Н). Соотношение между ними в отдельных природных объектах несколько колеблется, но более или менее близко к 6700:1, т. е. один атом 2Н (дейтерия) приходится примерно на 6700 атомов 1Н (протия). В ничтожных количествах — порядка одного атома на 1018 атомов 1Н — к ним примешан радиоактивный изотоп водорода 3Н (тритий), средняя продолжительность жизни атомов которого составляет 18 лет. Благодаря резкому количественному преобладанию протия над двумя другими изотопами природный водород может в первом приближении считаться состоящим из атомов 1Н.
Если говорить не только о земной коре, а о Вселенной в целом, то водород является самым распространённым элементом. На его долю приходится около 80% массы Юпитера и около 60% массы Сатурна. В межзвездном пространстве атомы водорода встречаются в несколько раз чаще, чем атомы всех остальных элементов, вместе взятых. Он резко преобладает над другими элементами также в атмосфере звёзд и, в частности, является главной составной частью солнечной атмосферы. Основной состав последней может быть выражен следующими данными (в атомных процентах):
| H | He | O | Mg | N | Si | S | C | Fe | Ca | Na | Ni | Al |
| 81,75 | 18,17 | 0,03 | 0,02 | 0,01 | 0,006 | 0,003 | 0,003 | 0,0008 | 0,0033 | 0,0003 | 0,0002 | 0,0002 |
Температура поверхности Солнца составляет около 5500 °С.
Основной формой существования водорода в космическом пространстве являются отдельные атомы Н. Ионизация их (по схеме Н = Н+ + е-) имеет определённое значение для теплового баланса этого пространства. Возникает она (как и диссоциация молекулы Н2 на атомы) в основном за счёт лучистой энергии звёзд, а при обратных процессах рекомбинации (Н+ + е- = Н и Н + Н = Н2) энергия выделяется главным образом в форме кинетической. Результатом является некоторое повышение температуры “околозвёздных” областей космического пространства по сравнению с очень далёкими от звёзд.
Создаваемая за счёт излучения кинетическая температура очень велика. Так, при длине волны 500 нм (что соответствует приблизительно середине видимого спектра) для её энергии имеем, что воспринимающая данное излучение частица обладает такой кинетической энергией, какую она имела бы при Т = 20 000 град.
Образование в естественных условиях, получение и применение. В природе водород образуется главным образом при разложении органических веществ, например целлюлозы или белков, некоторыми видами бактерий. Большие его количества освобождаются при коксовании угля; поэтому светильный и коксовый газы в среднем состоят на 50 объёмн. % из свободного водорода. В последнее время коксовый газ стали технически перерабатывать на водород, сжижая его и выделяя водород как трудно конденсирующийся газ.
В остальных случаях для получения водорода почти исключительно используют воду.
В настоящее время водород получают в огромных количествах. Очень большую часть его используют при синтезе аммиака, гидрогенизации жиров и при гидрировании угля, масел и углеводородов. Кроме того, водород применяют для синтеза соляной кислоты, метилового спирта, синильной кислоты, при сварке и ковке металлов, а также при изготовлении ламп накаливания и драгоценных камней. В продажу водород поступает в баллонах под давлением свыше 150 атм. Они окрашены в тёмно-зелёный цвет и снабжаются красной надписью “Водород”.
Большое техническое значение имеют следующие методы получения водорода (или азотно-водородной смеси): из водяного газа путём конверсии СО (контактный способ получения водяного газа), из природного или коксового газа в результате “расщепления метана”, из коксового или водяного газа фракционным сжижением, электролизом воды и железо-паровым способом. В качестве важнейшего побочного продукта водород получается в процессе электролиза водных растворов хлоридов щелочных металлов и при дуговом способе получения ацетилена. В ограниченном масштабе применяют также способ взаимодействия водяного пара с фосфором и термического разложения углеводородов:
СН4 (1000 °С) ® С + 2 Н2.
В некоторых случаях водород получают в результате каталитического расщепления метанола с водяным паром
СН3ОН + Н2О (250 °С) ® СО2 + 3 Н2,
или в результате каталитического термического разложения аммиака
2 NH3 (950 °С) ® N2 + 3 H2.
Однако эти исходные соединения получают в больших масштабах из водорода; между тем получение из них водорода является особенно простым и может быть использовано в таких производствах, которые потребляют его в сравнительно малых количествах (менее 500 м3/сутки).
Важнейшие методы получения водорода.
1. Растворение цинка в разбавленной соляной кислоте
Zn + 2 HCl = ZnCl2 + H2.
Этот способ чаще всего применяют в лабораториях.
Вместо соляной кислоты можно также использовать разбавленную серную кислоту; однако если концентрация последней слишком высока, то выделяющийся газ легко загрязняется SO2 и H2S. При использовании не вполне чистого цинка образуются ещё и другие соединения, загрязняющие водород, например AsH3 и PH3. Их присутствие и обусловливает неприятный запах получаемого этим способом водорода.
Для очистки водород пропускают через подкисленный раствор перманганата или бихромата калия, а затем через раствор едкого кали, а также через концентрированную серную кислоту или через слой силикагеля для освобождения от влаги. Мельчайшие капельки жидкости, захваченные водородом при его получении и заключённые в пузырьках газа, лучше всего устранять при помощи фильтра из плотно спрессованной обычной или стеклянной ваты.
Если приходится пользоваться чистым цинком, то к кислоте необходимо добавить две капли платинохлористоводородной кислоты или сернокислой меди, иначе цинк не вступает в реакцию.
2.Растворение алюминия или кремния в едкой щёлочи
2 Al + 2 NaOH + 6 H2O = 2 Na[Al(OH)4] + 3 H2
Si + 2 KOH + H2O = Na2SiO3 + 2 H2.
Эти реакции применяли раньше для получения водорода в полевых условиях (для наполнения аэростатов). Для получения 1 м3 водорода (при 0 °С и 760 мм рт. ст.) требуется только 0,81 кг алюминия или 0,63 кг кремния по сравнению с 2,9 кг цинка или 2,5 кг железа.
Вместо кремния также применяют ферросилиций (кремниевый метод). Смесь ферросилиция и раствора едкого натра, введённая в употребление незадолго до первой мировой войны во французской армии под названием гидрогенита, обладает свойством после поджигания тлеть с энергичным выделением водорода по следующей реакции:
Si + Ca(OH)2 + 2 NaOH = Na2SiO3 + CaO + 2 H2.
3. Действие натрия на воду
2 Na + 2 H2O = 2 NaOH + H2.
Ввиду того, что чистый натрий реагирует в этом случае слишком энергично, его чаще вводят в реакцию в виде амальгамы натрия; этот способ применяют преимущественно для получения водорода, когда им пользуются для восстановления “in statu nascendi”. Аналогично натрию с водой реагируют и остальные щелочные и щелочноземельные металлы.
4. Действие гидрида кальция на воду
СaН2 + 2 H2O = Сa(OH)2 + 2 H2.
Этот метод является удобным способом получения водорода в полевых условиях. Для получения 1 м3 водорода теоретически необходимо 0,94 кг СаН2 и, кроме воды, не требуется никаких других реактивов.
5. Пропускание водяного пара над раскалённым докрасна железом
4 Н2О + 3 Fe = Fe3O4 + 4 H2.
При помощи этой реакции в 1783 г. Лавуазье впервые аналитически доказал состав воды. Образующийся при этой реакции оксид железа нетрудно восстановить до металлического железа, пропуская над ним генераторный газ так, что пропускание водяного пара над одним и тем же железом можно провести произвольное число раз. Этот метод долгое время имел большое промышленное значение. В небольших масштабах его применяют и в настоящее время.
6. Пропускание водяного пара над коксом.
При температуре выше 1000 °С реакция идёт главным образом по уравнению
Н2О + С = СО + Н2.
Вначале получают водяной газ, т. е. смесь водорода и монооксида углерода с примесью небольших количеств углекислого газа и азота. От углекислого газа легко освобождаются промыванием водой под давлением. Монооксид углерода и азот удаляют при помощи процесса Франка-Каро-Линде, т. е. сжижением этих примесей, что достигается охлаждением жидким воздухом до -200 °С. Следы СО удаляют, пропуская газ над нагретой натронной известью
СО + NaOH = HCOONa — формиат натрия.
Этот метод даёт очень чистый водород, который используют, например, для гидрогенизации жиров.
Чаще, однако, водяной газ в смеси с парами воды при температуре 400 °С пропускают над соответствующими катализаторами, например над оксидом железа или кобальта (контактный способ получения водяного газа). В этом случае СО реагирует с водой по уравнению
СО + Н2Опар = СО2 + Н2 (“конверсия СО”).
Образующийся при этом СО2 поглощается водой (под давлением). Остаток монооксида углерода (~1 об. %) вымывают аммиачным раствором однохлористой меди. Применяемый в этом способе водяной газ получают пропусканием водяного пара над раскалённым коксом. В последнее время всё больше используют взаимодействие водяного пара с пылевидным углём (превращение угольной пыли в газы). Полученный таким способом водяной газ содержит обычно большое количество водорода. Выделяемый из водяного газа водород (содержащий азот) применяют главным образом для синтеза аммиака и гидрирования угля.
7. Фракционное сжиженнее коксового газа.
Подобно получению из водяного газа, водород можно получать фракционным сжижением коксового газа, основной составной частью которого является водород.
Сначала коксовый газ, из которого предварительно удаляют серу, очищают от СО2 промыванием водой под давлением с последующей обработкой раствором едкого натра. Затем постепенно освобождают от остальных примесей ступенчатой конденсацией, проводимой до тех пор, пока не остаётся только водород; от других примесей его очищают промыванием сильно охлаждённым жидким азотом. Этот метод применяют главным образом, чтобы получить водород для синтеза аммиака.
8. Взаимодействие метана с водяным паром (разложение метана).
Метан взаимодействует с водяным паром в присутствии соответствующих катализаторов при нагревании (1100 °С) по уравнению
СН4 + Н2Опар + 204 кДж (при постоянном давлении).
Необходимое для реакции тепло следует подводить или извне, или применяя “внутреннее сгорание”, т. е. подмешивая воздух или кислород таким образом, чтобы часть метана сгорала до диоксида углерода
СН4 + 2 О2 = СО2 + 2 Н2Опар + 802 кДж (при постоянном давлении).
При этом соотношение компонентов выбирают с таким расчётом, чтобы реакция в целом была экзотермичной
12 СН4 + 5 Н2Опар + 5 О2 = 29 Н2 + 9 СО + 3 СО2 + 85,3 кДж.
Из монооксида углерода посредством “конверсии СО” также получают водород. Удаление диоксида углерода производят вымыванием водой под давлением. Получаемый методом разложения метана водород используют главным образом при синтезе аммиака и гидрировании угля.
9. Взаимодействие водяного пара с фосфором (фиолетовым).
2 Р + 8 Н2О = 2 Н3РО4 + 5 Н2.
Обычно процесс проводят таким образом: пары фосфора, получающиеся при восстановлении фосфата кальция в электрической печи, пропускают вместе с водяным паром над катализатором при 400-600 °С (с повышением температуры равновесие данной реакции смещается влево). Взаимодействие образовавшейся вначале Н3РО4 с фосфором с образованием Н3РО3 и РН3 предотвращают быстрым охлаждением продуктов реакции (закалка). Этот метод применяют прежде всего, если водород идёт для синтеза аммиака, который затем перерабатывают на важное, не содержащее примесей удобрение — аммофос (смесь гидро- и дигидрофосфата аммония).
10. Электролитическое разложение воды.
Чистая вода практически не проводит тока, поэтому к ней прибавляются электролиты (обычно КОН). При электролизе водород выделяется на катоде. На аноде выделяется эквивалентное количество кислорода, который, следовательно, в этом методе является побочным продуктом.
Получающийся при электролизе водород очень чист, если не считать примеси небольших количеств кислорода, который легко удалить пропусканием газа над подходящими катализаторами, например над слегка нагретым палладированным асбестом. Поэтому его используют как для гидрогенизации жиров, так и для других процессов каталитического гидрирования. Водород, получаемый этим методом довольно дорог.
Физические свойства. Водород (т. пл. -259, т. кип. -253 °С) — бесцветный газ, не имеющий запаха. В воде он растворяется незначительно — 2:100 по объёму. Для него характерна растворимость в некоторых металлах.
В отличие от прочих газов (кроме гелия), водород самопроизвольно расширяется при обычных температурах не с охлаждением, а с разогреванием. Он начинает вести себя “нормально” лишь ниже -80 °С.
Жидкий водород имеет плотность около 0,07 г/см3, твёрдый — около 0,08 г/см3. Теплота его плавления 117 Дж/моль, а испарения — 915 Дж/моль. Критическая температура водорода -240 °С, а критическое давление 13 атм.
Распад молекулы водорода на атома требует большой затраты энергии — 436 кДж/моль при 25 °С. Ионизации может подвергаться и молекула Н2. Процесс идёт с образованием положительно заряженного “молекулярного иона”:
Н2 + 1492 кДж = Н2+ + е-..
В ионе Н2+ (d(HH) = 106 пм) между обоими частицами осуществляется одноэлектронная связь. Последняя значительно менее прочна (энергия разрыва 259 кДж/моль), чем обычная двухэлектронная связь в нейтральной молекуле Н2.
Хорошо растворяют водород, в частности, Ni, Pt и Pd, причём один объём палладия может поглотить несколько сотен объёмов водорода. Наоборот, некоторые другие металлы (например, Ag) его практически не растворяют. С растворением водорода в меди и железе приходится считаться при отливке из них изделий, так как взаимодействие этого газа с присутствующими в металле следами оксидов ведёт к образованию водяного пара, который вызывает возникновения в литье трещин и пустот. Вместе с тем способность водорода проходить сквозь нагретые металлические части аппаратуры создаёт большие технические трудности работы с ним при высоких температурах и давлениях.
Так как водород является самым легким из газов, молекулы его движутся быстрее остальных. Поэтому водород характеризуется наибольшей скоростью диффузии, т. е. скорее других газов распространяется в пространстве, проходит сквозь различные мелкие поры и т. д. Этим же обусловлена и его высокая теплопроводность. Так, при прочих равных условиях нагретый предмет охлаждается водородом в семь раз быстрее, чем воздухом.
Химическая роль водорода весьма многообразна, и его производные — гидриды — известны для многих элементов. Атом водорода может либо отдавать свой единственный электрон с образованием положительного иона (представляющего собой голый протон), либо присоединять один электрон, переходя в отрицательный ион, имеющий гелиевую электронную конфигурацию.
Полный отрыв электрона от атома водорода требует затраты очень большой энергии ионизации:
H + 1400 кДж = H+ + е-
Вследствие этого при взаимодействии водорода с неметаллами возникают не ионные, а лишь полярные связи.
Тенденция того или иного нейтрального атома к присоединению избыточного электрона характеризуется значением его сродства к электрону. У водорода оно выражено довольно слабо:
Н + е-= Н- + 79 кДж
Несмотря на это, ионные структуры, содержащие в своём составе Н- известны. Соединения такого типа образуются прямым взаимодействием наиболее активных металлов (Na, Ca и др.) c водородом при нагревании. По своему характеру они являются типичными солями, похожими на соответствующие производные фтора и хлора. Однако из-за их неустойчивости по отношению к воде и воздуху иметь с ними дело приходится сравнительно редко.
Образование иона Н- (по схеме Н + е- = Н- + hv) играет значительную роль в процессе возникновения солнечного излучения. Не исключена возможность их промежуточного образования в процессе взаимодействия металлов с кислотами (по схемам: Zn + H+ = Zn2+ + H- и затем H- + H+ = H2).
По типу более или менее полярной связи водород соединяется со многими неметаллами: кислородом, хлором, серой, азотом и др. В их рациональных названиях для атома водорода применяют термин “гидро” или “ацидо”.
Водород не поддерживает горение обычных горючих веществ (являющихся соединениями углерода). Так, зажжённая свеча гаснет в нём. Однако, например, кислород горит в атмосфере водорода. Отсюда видна относительность понятия “поддерживает” или “не поддерживает” горения. Обычно его относят именно к горению соединений углерода.
Сам водород горит и в чистом кислороде, и на воздухе, причём продуктом сгорания является вода. При поджигании смеси обоих газов (“гремучего газа”) взаимодействие протекает со взрывом. Если вместо поджигания привести эту смесь в соприкосновение с очень малым количеством мелко раздробленной платины (играющей роль катализатора), то реакция протекает быстро, но спокойно.
Реакция образования воды из водорода и кислорода сильно экзотермична:
2 Н2 + О2 = 2 Н2О + 573 кДж
Помимо прямого соединения с кислородом водород способен отнимать его от оксидов многих элементов: Cu, Pb, Hg и др. В результате из оксида получается свободный элемент, например:
СuO + H2 = H2O + Cu + 130 кДж.
Однако эти реакции, в которых водород выступает как восстановитель, протекают лишь при нагревании. При высоких давлениях водород вытесняет некоторые металлы также из растворов их солей.
Опыт показывает, что химическая активность водорода иногда сильно повышается. Это наблюдается тогда, когда реагирующие с ним вещества находятся в непосредственном контакте с выделяющимся водородом. Повышенную активность такого водорода “в момент выделения” (“in statu nascendi”) объясняется тем, что реагируют не молекулы Н2, а атомы. Действительно, при реакциях получения водорода (например, действием цинка на кислоту) первоначально выделяются именно отдельные атомы. Если же у места их выделения имеется вещество, способное с ними реагировать, то такая реакция может происходить без предварительного образования молекул Н2.
Это представление было косвенно подтверждено, когда удалось получить атомарный водород в газообразном состоянии и изучить его реакционную способность. Оказалось, что он значительно активнее молекулярного. Так, атомарный водород уже при обычных условиях соединяется с серой, фосфором, мышьяком и т. д., восстанавливает оксиды многих металлов, вытесняет некоторые металлы (Cu, Pb, Ag и др.) из их солей и вступает в другие химические реакции, на которые при тех же условиях не способен обычный молекулярный водород.
При химических взаимодействиях с участием обычного водорода молекула его должна распадаться на атомы. Но сама реакция такого распада (диссоциация на атомы) сильно эндотермична:
Н2 + 435 кДж = Н + Н.
Очевидно, что затрачиваемая на эту реакцию энергия (энергия диссоциации) должна быть восполнена энергией, выделяющуюся при взаимодействии атомов водорода с введённым в реакцию веществом. Следовательно, можно ожидать, что реакция водорода, при которых выделяется менее 435 кДж/моль, не будет протекать самопроизвольно. В случае взаимодействия веществ с атомарным водородом такой затраты энергии на диссоциацию уже не требуется. Поэтому здесь и возможен значительно более широкий круг реакций.
Атомарный водород удобно получать действием на обычный водород тихого электрического разряда. При этом часть молекул распадается на атомы, которые под уменьшенным давлением соединяются в молекулы не моментально, благодаря чему и могут быть изучены химические свойства атомарного водорода.
Аналогично водороду может быть получен в атомарном состоянии и кислород. Его химическая активность при переходе в атомарное состояние тоже резко возрастает.
Большое количество энергии, выделяющейся при образовании молекулы водорода, объясняет её устойчивость при обычных условиях. Вместе с тем оно же наводит на мысль о возможности термической диссоциации (разложения при нагревании) молекулы Н2, если сообщить ей достаточное количество тепла. Опыт показывает, что заметная термическая диссоциация водорода начинается примерно с 2000 °С и происходит тем в большей степени, чем выше температура. Наоборот, при понижении температуры отдельные атомы вновь соединяются в молекулы.
Термическая диссоциация водорода (под обычным давлением) характеризуется следующими данными:
| Абсолютная температура, К | 2000 | 2500 | 3000 | 3500 | 4000 | 5000 |
| Диссоциированная часть, % | 0,088 | 1,31 | 8,34 | 29,6 | 63,9 | 95,8 |
Переход водорода в атомарное состояние может вызываться также излучением с длинами волн менее 85 нм. Этим и обусловлено резкое преобладание атомарного водорода над молекулярным в космическом пространстве.
Соединение атомов водорода в молекулы протекает значительно быстрее на поверхности металлов, чем в самом газе. При этом металл воспринимает ту энергию, которая выделяется при образовании молекул и нагревании до очень высоких температур. Последнее создаёт возможность технического использования атомарного водорода для атомно-водородной сварки металлов: между двумя вольфрамовыми стержнями создаётся электрическая дуга, сквозь которую по облегающим стержни трубкам пропускается ток водорода. При этом часть молекул Н2 распадается на атомы, которые затем вновь соединяются на металлической поверхности, помещенной недалеко от дуги. Таким путём металл может быть нагрет выше 3500 °С. В этих условиях происходит быстрая и прочная сварка отдельных его кусков. Большим достоинством атомно-водородной сварки является равномерность нагрева, позволяющая сваривать даже тонкие металлические детали.
Соединение атомов водорода осуществляется гораздо легче на твёрдой поверхности. При реакции по схеме Н + Н = Н2 молекула водорода заключает в себе и кинетическую энергию обоих соединяющихся атомов, и энергию их взаимодействия. В сумме это даёт запас энергии, с избытком превышающий энергию диссоциации молекулы Н2 на атомы. Такая диссоциация не происходит только в том случае, если молекула быстро освобождается от избытка энергии, передавая его какой-либо другой частице. В самом газе это может осуществляться лишь путём тройного столкновения по схеме Н + Н + Х ® Н2 + Х, где Х — частица, принимающая избыток энергии. Но вероятность тройного столкновения несравненно меньше вероятности двойного, и поэтому в газе рекомбинация (обратное соединение) атомов Н идёт сравнительно медленно. Напротив, у твёрдой поверхности к образованию молекулы может вести каждое двойное столкновение атомов Н, так как воспринимающая избыток энергии частица (в виде атома или молекулы вещества самой поверхности) всегда имеется.
Если в колбу электрической лампы ввести водород (вместо аргона), то около раскалённой вольфрамовой нити будут происходить частичная диссоциация молекул Н2 на атомы. Энергия рекомбинации последних на покрытой специальным составом (люминофором) внутренней поверхности колбы вызывает её интенсивное свечение. Было показано, что от таких ламп при равной мощности можно получить значительно больше света, чем от обычных.
Практическое применение водорода многообразно: им обычно заполняют шары-зонды, в химической промышленности он служит сырьём для получения многих весьма важных продуктов (аммиака и др.), в пищевой — для выработки из растительных масел твёрдых жиров и т. д. Высокая температура (до 2600 °С), получающаяся при горении водорода в кислороде, используется для плавления тугоплавких металлов, кварца и т. п. Жидкий водород является одним из наиболее эффективных реактивных топлив. Ежегодное мировое потребление водорода превышает 1 млн. т. Эффективность жидкого водорода как реактивного топлива гораздо выше, чем спирта или керосина. Так, в комбинации с кислородом он может дать удельный импульс 390 с.
Вода.
Природные воды всегда содержат примеси. Одни из них находятся во взвешенном состоянии, другие — в растворённом. От большей части взвешенных частиц вода может быть освобождена отстаиванием или, быстрее, фильтрованием сквозь толстые слои песка и т. п. В лаборатории для этой цели применяется фильтровальная (непроклеенная) бумага. От растворённых веществ воду обычно очищают перегонкой. Такая перегнанная вода называется дистиллированной.
Обычно применяемая в городском хозяйстве схема очистки речной воды состоит из нескольких стадий: первой операцией является добавка к воде небольшого количества сернокислого алюминия, который выделяет объёмный осадок гидроксида алюминия, захватывающий различные взвешенные в воде частицы и тем способствующий их последующему осаждению в отстойнике. Отстоявшаяся вода фильтруется сквозь толстый слой песка, затем обеззараживается хлорированием и лишь после этого поступает в водопроводную сеть (для Москвы 10 млн. м3 ежедневно).
Большие преимущества перед хлорированием во многих случаях имеет стерилизация воды путём её озонирования. Технически этот процесс вполне освоен, но обходится он в несколько раз дороже, что и затрудняет его широкое внедрение.
Перегнанная вода свободна только от нелетучих примесей. От летучих её стараются освободить, добавляя перед перегонкой вещества, реагирующие с этими примесями и дающие с ними нелетучие продукты реакции. Всё же и тогда первые порции перегоняемой воды содержат растворённые газы воздуха. В тех случаях, когда их присутствие вредит, эти порции не собирают.
Как при самой перегонке в стеклянных сосудах, так и при хранении в них дистиллированная вода загрязняется переходящими в неё из стекла щелочами. Громадному большинству её применений эти растворённые щелочи не вредят, так как их ничтожно мало. Если требуется ещё более высокая чистота, то дистиллированную воду получают и сохраняют в сосудах из кварца, олова и серебра.
Вода состоит из 11,2 вес. % водорода и 88,8 вес. % кислорода. При образовании её из элементов с одним объёмом кислорода соединяются два объёма водорода. В том же, конечно, соотношении находятся объёмы газов получаемых при разложении воды на элементы.
Термическая диссоциация водяного пара протекает по двум параллельным реакциям:
2 Н2О + 485 кДж = 2 Н2 + О2 и 2 Н2О + 564 кДж = 2 Н2 + 2 ОН.
Первоначально преобладает первая из них, и лишь при очень высоких температурах начинает преобладать вторая.
Образующиеся в результате протекания этой реакции гидроксильные радикалы термически весьма устойчивы. Так, было установлено наличие их в атмосфере Солнца.
Оба атома водорода в молекуле воды расположены по одну сторону от атома кислорода. Вследствие этого и высокой полярности связей Н-О молекула воды характеризуется высокой полярностью, РНОН = 104,5°, d(HO) = 96 пм.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Молекула воды имеет эффективный радиус 138 пм (из кристаллической структуры льда). Её общая полярность имеет значение m = 1,88, причём структура дипольного момента не вполне ясна. Если допустить, что он обусловлен не только полярностью связей Н-О, но и орбиталями занимаемыми электронными парами атома кислорода, то молекула воды при равномерном распределении эффективных зарядов между вершинами тетраэдров каждая из них должна иметь d = ±0,17.
Молекула воды имеет эффективный радиус 138 пм (из кристаллической структуры льда). Её общая полярность имеет значение m = 1,88, причём структура дипольного момента не вполне ясна. Если допустить, что он обусловлен не только полярностью связей Н-О, но и орбиталями занимаемыми электронными парами атома кислорода, то молекула воды при равномерном распределении эффективных зарядов между вершинами тетраэдров каждая из них должна иметь d = ±0,17.
Результаты определения молекулярного веса водяного пара указывают на то, что ему соответствует простейшая формула — Н2О. Наоборот, в жидком состоянии вода ассоциирована, т. е. наряду с простыми молекулами содержит и более сложные образования, соответствующие общей формуле (Н2О)n, где n = 2, 3, 4 и т. д. Подобные молекулярные агрегаты всё время возникают и вновь распадаются, что можно выразить схемой: nH2O Ы (H2O)n. При нагревании воды степень её ассоциации уменьшается.
Определение плотности водяного пара при температуре кипения воды даёт для молекулярного веса значение 18,64, что соответствует наличию в паре около 3,5 % удвоенных молекул. Существование в виде подобных молекул (Н2О)2 довольно характерно для воды, растворённой в некоторых органических жидкостях (например, хлороформе).
Вообще говоря, причиной ассоциации молекул может быть их высокая полярность. Молекулы воды, являющиеся постоянными диполями, могут за счёт взаимного притяжения противоположно заряженных полюсов комбинироваться по две, три и т. д. Однако действующие при этом силы притяжения невелики, и в случае воды подобная дипольная ассоциация играет лишь второстепенную роль.
Основное значение для ассоциации молекул воды имеет образование так называемых водородный связей. Последние возникают за счёт притяжения водорода одной молекулы воды к кислороду другой по приводимой схеме:
Н
Ѕ
О—Н···О—Н
Ѕ
Н
Возможность такого притяжения согласуется с допущением о наличии значительных эффективных зарядов и у водорода (dН = +0,33), и у кислорода (dО = -0,66) в молекуле воды.
Так как при этом первоначальная связь водорода со своим “собственным” кислородом не теряется, он оказывается одновременно связанным с двумя кислородами и тем самым связывает обе молекулы воды друг с другом. Подобным же образом, за счёт образования водородных связей могут стянуться вместе три, четыре и более молекул воды. По-видимому, в жидкой воде каждая из них связывается с другими, в среднем, двумя Н-связями. Прочность водородной связи гораздо меньше, чем обычных валентных (энергия её для воды составляет примерно 21 кДж/моль). Поэтому связанные вместе молекулы могут разойтись, затем вновь связаться в других комбинациях и т. д.
Необходимым условием возникновения Н-связей является достаточная полярность валентных связей водорода в исходных молекулах. Так как этому более всего удовлетворяют связи Н-F, H-O и H-N, среди содержащих их соединений обычно и встречаются вещества, для которых характерно ассоциация за счёт образования Н-связей. Последние могут возникать и между не одинаковыми молекулами (например, воды и спирта).
Жидкая вода в тонких слоях бесцветна, а в толстых имеет голубовато-зелёный цвет. В противоположность почти всем другим веществам, плотность которых по мере охлаждения всё время возрастает, вода имеет наибольшую плотность при +4 °С.
Причина плотностной аномалии воды точно не установлена. Предполагают, что при 0 °С вода в значительной части состоит из (Н2О)3, а при нагревании её до +4 °С утроенные молекулы переходят в (Н2О)2, что сопровождается увеличением плотности. При дальнейшем нагревании начинают преобладать простые молекулы, и плотность постепенно уменьшается. Другое объяснение плотностной аномалии воды допускает существование в ней при низких температурах мельчайших кристаллов льда. Предполагается, что при 0 °С вода содержит 0,6 % таких кристалликов, а с повышением температуры количество их очень быстро уменьшается. Наконец, третье возможное объяснение этого явления исходит из наличия некоторой упорядоченности в структуре жидкостей. Предполагается, что при нагревании от 0 до 4 °С характер этой упорядоченности у воды изменяется таким образом, что результатом является более тесное сближение частиц.
Повышение давления смещает максимальную плотность воды в сторону более низких температур. Так, при 50 атм максимальная плотность наблюдается около 0 °С выше 2000 атм аномалия плотности воды исчезает.
Чистая вода почти не проводит электрический ток. Она характеризуется наибольшей их всех жидких и твёрдых веществ удельной теплоемкостью, т. е. для нагревания воды требуется затратить больше тепла, чем для нагревания на тоже число градусов равного по массе количества какой-либо другой жидкости или твёрдого тела. Обратно, при охлаждении вода отдает больше тепла, чем равное количество любого твёрдого или жидкого вещества.
Как и плотность, теплоемкость воды меняется с температурой аномально. В противоположность обычно наблюдающемуся последовательному увеличению теплоемкости, для воды она сначала падает, а лишь затем вновь начинает возрастать.
теплоемкость воды значительно больше, чем у других жидкостей (кроме металлов), и изменяется тоже аномально: до 150 °С возрастает, и лишь затем начитает уменьшаться. Электропроводность воды очень мала, но заметно возрастает при повышении и температуры, и давления. Критическая температура воды равна 374 °С, критическое давление 218 атм.
Быстро уменьшается при нагревании вязкость воды. Поэтому горячие растворы фильтруются значительно быстрее холодных. Интересно, что при сравнительно низких температурах (примерно до 20 °С) зависимость вязкости воды от давления около 1000 атм проходит через минимум, который при более высоких температурах не наблюдается. Растворимые соли, как правило, повышают вязкость воды.
\Показатель преломления воды на протяжении видимого спектра почти не изменяется (1,33 для красных лучей и 1,34 для фиолетовых при 20 °С). С повышением температуры он несколько уменьшается, а с повышением давления возрастает. Инфракрасные лучи поглощаются водой очень сильно, тогда как для ультрафиолетовых она довольно прозрачна.
Скорость распространения звука в воде (около 1400 м/с при 4 °С) примерно в 4 раза больше, чем в воздухе. По мере нагревания воды до 80 °С она несколько возрастает, а затем начинает уменьшаться.
Так как молекулы воды сильно притягиваются друг к другу, она характеризуется большой величиной поверхностного натяжения. Расположенная внутри жидкости молекула находится под действием притяжения соседних частиц одинаково со всех сторон. Напротив, лежащая на поверхности молекула испытывает притяжение только с нижней стороны и тем самым втягивается внутрь жидкости. Поэтому и вся поверхность находится в состоянии известного натяжения.
Под воздействием поверхностного натяжения небольшие количества воды стремятся принять шарообразную форму, соответствующую наименьшей возможной величине поверхности для данного количества вещества. Приближение к форме шара достигается тем большее, чем слабее сказывается сила тяжести, т. е. чем меньше вес капли. Таким образом, форма очень маленькой капельки воды близка к точно шарообразной. Следует отметить, что поверхностное натяжение воды очень чувствительно даже к следам примесей.
При соприкосновении жидкости с каким-либо нерастворимым в ней твёрдым веществом, например стеклом, могут быть два случая. Если притяжение молекул жидкости к молекулам твёрдого вещества сильнее, чем друг к другу, мениск (т. е. поверхность раздела с воздухом) находящейся в стеклянной трубке жидкости будет вогнутым, в противном случае — выпуклым. Первое наблюдается, например, у воды, второе — у ртути. Обычно говорят, что вода “смачивает” стекло, а ртуть “не смачивает”. Если внутреннюю поверхность стеклянной трубки покрыть парафином, то вода не будет её смачивать и форма мениска станет выпуклой.
При низких температурах вода испаряется сравнительно медленно, но при нагревании давление её пара быстро возрастает:
Температура, °С | 5 | 10 | 15 | 20 | 25 | 30 | 40 | 50 | 75 | 100 | |
| Давление пара, кПа | 0,61 | 0,87 | 1,23 | 1,71 | 2,33 | 3,17 | 4,24 | 7,37 | 12,3 | 38,5 | 101 |
Если в каком-нибудь замкнутом пространстве над жидкой водой находится воздух, то парциальное давление в нём водяного пара соответствует приведённым значениям. Такой воздух будет насыщен водяным паром — больше содержаться в нём последнего при данной температуре не может. Обычно воздух содержит от 30 до 90 % максимально возможного количества водяного пара.
Абсолютное содержание водяного пара в насыщенном им воздухе изменяется с температурой следующим образом:
Температура, °С | -20 | -10 | +10 | 20 | 30 | |
Содержание водяного пара, г/м3 | 1,08 | 2,35 | 4,85 | 9,41 | 17,3 | 30,4 |
Под относительной влажностью воздуха понимается выраженное в процентах отношение действительного содержания водяных паров к отвечающему состоянию насыщения при данной температуре. Наиболее благоприятные для человеческого организма условия относительной влажности применительно к обычным комнатным температурам (t) хорошо передаются формулой 50 - 3·(t - 20). Как видно из последней, чем выше температура, тем меньше должна быть относительная влажность.
Относительная влажность воздуха зависит от географического положения местности (и многих других факторов). Например, для Москвы её усреднённые значения — минимальное (август), максимальное (февраль) и среднегодовое — равны соответственно 57, 85 и 72 %.
Для поддержания определённой влажности воздуха в закрытых помещениях иногда используются насыщенные растворы соответственно подобранных солей. Например, относительная влажность около 50 % при 20 °С может быть поддерживаема с помощью Са(NO3)2 или NaHSO4.
При охлаждении ненасыщенного водяным паром воздуха постепенно достигается состояние насыщения, после чего избыточный водяной пар начинает выделяться в виде тумана или — при резком охлаждении — в виде дождя. Если весь процесс проходит при более низких температурах, получается соответственно иней и снег.
Когда давление пара в жидкости становится равным внешнему давлению, она закипает. Для воды под нормальным атмосферным давлением (101325 Па) температура кипения равна 100 °С. Очевидно, что при уменьшении давления эта температура будет понижаться, при увеличении — повышаться. Некоторые данные для близких к нормальному и высоких давлений сопоставлены ниже:
| Атмосферное давление, кПа | 97,3 | 98,7 | 100 | 101,3 | 102,7 | 104,0 |
Температура кипения, °С | 98,9 | 99,3 | 99,6 | 100,0 | 100,4 | 100,7 |
| Давление, атм | 2 | 5 | 10 | 20 | 50 | 100 |
Температура кипения, °С | 120 | 151 | 179 | 211 | 263 | 310 |
Приведённые данные показывают, что по мере роста давления температура кипения воды повышается очень быстро.
Если воду тщательно освободить от взвешенных частиц и растворённых газов, и затем равномерно нагревать, предохраняя от встряхивания, то может быть достигнута температура значительно выше 100 °С, прежде чем вода бурно вскипит. При перемешивании такой перегретой воды вскипание обычно происходит тотчас же. Практически удавалось доводить перегрев воды почти до 270 °С. Последняя температура является, по-видимому, предельной для возможного перегрева воды под обычным давлением.
Со сравнительно небольшим перегревом часто приходится встречаться при кипячении жидкости, которые в этом случае кипят “толчками”. Для устранения перегрева и связанных с ним явлений в жидкость иногда вводят запаянные с одного конца очень тонкие (“капиллярные”) стеклянные трубки, так как задерживающийся в них воздух способствует равномерности кипения.
Для перевода веществ из жидкого в газообразное состояние необходимо затратить работу на преодоление взаимного притяжения молекул и внешнего давления. Величина этой работы, выражена в джоулях, называется теплотой испарения данного вещества. Последняя зависит от температуры, при которой происходит испарение, причём уменьшается по мере её повышения и при критической температуре становится равной нулю. Для воды при 100 °С имеем: Н2О(ж) + 41 кДж = Н2О(г). При переходе пара в жидкость это же количество тепла выделяется. Кипящая вода не может быть под атмосферным давлением нагрета выше 100 °С, т. к. всё избыточно подводимое тепло тратится на испарение. Следует отметить, что из всех жидкостей вода характеризуется наибольшим значением теплоты испарения на единицу массы.
Деление теплоты испарения жидкости на её молярный объём (при той же температуре) приводит к значению внутреннего давления данной жидкости (Р), которое может служить мерой сил связи между её молекулами. Например, для воды при 100 °С молярный объём составляет 18,8 см3 и Р = 41:18,8 = 2,18 кДж/см3. Перевод этой величины в единицы давления при помощи механического эквивалента тепла даёт 22000 атм. Таким образом, внутреннее давление воды очень велико. Подавляющее большинство других жидкостей характеризуется внутренними давлениями порядка 2000-5000 атм, т. е. гораздо меньшими, чем у воды.
Из-за большой величины внутреннего давления сжимаемость воды мала. В то время как обычно сжимаемость жидкостей при повышении температуры возрастает, у воды она изменяется аномально, проходя около 50 °С через минимум, положение которого практически не зависит от давления. Растворённые соли существенно снижают сжимаемость воды.
Несмотря на свою небольшую величину, сжимаемость воды важна для жизни природы, т. к. снижает уровень мирового океана. Было подсчитано, что при отсутствии сжимаемости этот уровень стоял бы приблизительно на 30 метров выше современного (что привело бы к затоплению около 4 % всей площади суши).
При охлаждении воды до 0 °С она переходит в твёрдое состояние — лёд. Плотность льда равна 0,92 г/см3, т. е. он легче воды. Это обстоятельство имеет громадное значение для сохранения жизни, т. к. благодаря ему образующийся в водоёмах лёд остаётся на поверхности воды и предохраняет более глубокие её слои от дальнейшего охлаждения. Если бы лёд был тяжелее воды, все водоёмы холодного и умеренного поясов представляли бы собой массы льда, лишь в летнее время оттаивающие с поверхности. Свойство воды в данном случае аномальны, т. к. у громадного большинства веществ плотность в твёрдом состоянии больше, чем в жидком.
Если очень чистую воду охлаждать, предохраняя от сотрясений, то её можно переохладить, т. е. достигнуть температур ниже нуля без образования льда. Однако такая переохлаждённая вода малоустойчива — при внесении в неё кристаллика льда она затвердевает.
Особенно легко переохлаждаются отдельные капли воды, причём их самопроизвольное замерзание наступает тем труднее, чем они меньше. Так, при диаметрах от одного мм до одного мк температуры быстрого самопроизвольного замерзания водяных капель лежат в пределах от -24 до -38 °С. Поэтому облака даже при низких температурах состоят обычно не из частиц льда, а из капелек воды. Каждый см3 дождевого облака содержит от десятков до сотен капелек с диаметрами от 1 мк до 1 мм.
Некоторые растворённые в воде примеси существенно влияют на её способность к переохлаждению. Например, при небольшой добавке ацетона удавалось переохлаждать водяные капли до -72 °С. Подобные примеси имеются, вероятно, в крови холоднокровных животных, благодаря чему их организмы способны без вреда для себя переносить замораживание и последующее оттаивание. Напротив, у теплокровных животных способность крови к переохлаждению очень невелика. Происходящая при её замораживании кристаллизация воды вызывает разрывы тканей с их последующим омертвением.
При обычных условиях состояние жидкой воды является устойчивым. Напротив, переохлаждённая или перегретая вода находится в так называемом метастабильном состоянии. Последнее характеризуется тем, что само по себе оно более или менее устойчиво, но устойчивость эта легко нарушается под влиянием тех или иных воздействий. Если представить себе конус со слегка срезанной параллельно основанию вершиной, то устойчивое состояние вещества будет соответствовать такому конусу, стоящему на своём основании, а метастабильное — стоящему на вершине. Возможность более или менее длительного существования метастабильных состояний обусловлено затруднённостью возникновения при данных условиях зародышевых образований стабильной фазы рассматриваемого вещества.
Малая плотность льда связана с наличием значительных пустот в его кристаллической структуре. Последняя образована молекулами воды, соединёнными друг с другом Н-связями. Каждый атом кислорода связан с двумя “своими” атомами водорода [на расстоянии d(HO) = 100 пм] и двумя “чужими” [d(НО) = 176 пм]. Таким образом на каждую молекулу воды приходится четыре водородные связи (рис. ), которые обеспечивают устойчивость структуры льда. Схеме расположения кислородных атомов в этой структуре показана на рис., а атомы водорода располагаются вдоль соединительных линий.
![]() •
•
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() 176 пм
176 пм
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Н •
Н •
![]()
![]()
![]() 100 пм
100 пм
![]()
![]()
![]()
![]()
![]() О •
О •
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Н • •
Н • •
![]()
![]()
![]()
![]()
![]()
![]()
![]() • • •
• • •
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() •
•
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Рис. 1. Схема распределения Рис. 2. Структурная схема льда.
связей в кристалле льда.
![]()
![]() Подобно воде, испаряться может и лёд. Хорошо известно, например, что мокрое бельё сохнет даже при больших морозах. Установлено также, что за зимний период испаряется до 30% всего выпавшего снега. Однако испарение льда идёт гораздо медленнее, чем жидкой воды, т. к. давление водяного пара надо льдом при низких температурах весьма мало:
Подобно воде, испаряться может и лёд. Хорошо известно, например, что мокрое бельё сохнет даже при больших морозах. Установлено также, что за зимний период испаряется до 30% всего выпавшего снега. Однако испарение льда идёт гораздо медленнее, чем жидкой воды, т. к. давление водяного пара надо льдом при низких температурах весьма мало:
Температура, °С | -50 | -30 | -20 | -10 | -8 | -6 | -4 | -2 | |
| Давление пара, Па | 4 | 40 | 107 | 253 | 307 | 373 | 440 | 520 | 613 |
Испарение льда отнюдь не является исключением. Некоторое (обычно ничтожное и не поддающееся непосредственному измерению) давление пара имеется над любым твёрдым веществом. Иногда оно настолько велико, что становится заметным. Примером может служить нафталин, применяемый для предохранения одежды от моли.
Так как в равновесной системе вода Ы лёд объём льда больше объёма того же количества воды, можно ожидать, что при увеличении давления равновесие сместится влево. Практически это значит, что при высоких давлениях лёд будет плавиться ниже 0 °С. Действительно, опыт показывает, что каждая атмосфера избыточного давления понижает температуру плавления льда приблизительно на 0,008 град. Таким образом, смещение точки плавления весьма значительно. Обусловлено это тем, что объём льда лишь немногим больше объёма того же количества воды.
При плавлении льда температура не поднимается выше 0 °С, потому что всё избыточно сообщаемое извне тепло тратится на плавление (теплота плавления): Н2О(т) + 6 кДж = Н2О(ж). При замерзании воды это же количество тепла выделяется. Теплоёмкость льда гораздо меньше (примерно вдвое), а теплопроводность несколько больше, чем у жидкой воды.
Из изложенного выше вытекает, что при плавлении льда (или снега) без подведения тепла извне температура должна понижаться. В правильности этого вывода можно убедиться, облив небольшое количество снега спиртом: вследствие образования раствора происходит быстрое таяние снега, сопровождающееся сильным охлаждением жидкости. Так как при смешивании со спиртом жидкой воды происходит заметное разогревание смеси, наблюдающееся при растворении снега охлаждение обусловлено именно его плавлением.
Как видно из рис. 1, во льде число окружающих каждый атом кислорода на ближайшем к нему расстоянии таких же атомов равно четырём. С помощью рентгеновского анализа установлено, что плавление льда сопровождается повышением среднего значения этого числа до 4,4 (при 1,5 °С), а последующее нагревание воды — его дальнейшим увеличением (до 4,9 при 83 °С). Само по себе такое увеличение должно было бы вести к возрастанию плотности воды. Однако одновременно возрастает среднее расстояние между соседними атомами кислорода (от 276 пм во льде до 290 пм при 1,5 °С и 305 пм при 83 °С), что и приводит к снижению плотности.
В современной науке преобладает мнение, согласно которому плавление льда сопровождается не полным, а лишь частичным разрушением его кристаллической структуры, отдельные пустоты которой заполняются отдельными молекулами воды. С этой точки зрения, основная масса жидкой воды слагается при обычных условиях из менее или более разрыхлённой и искажённой кристаллической сетки льда, находящейся в состоянии непрерывной перестройки. В свете этих данных плотностную аномалию воды можно истолковать следующим образом: от 0 до 4 °С основное значение имеет повышение среднего числа окружающих каждую молекулу Н2О ближайших соседей, а при дальнейшем нагревании — увеличение среднего расстояния между ними.
По другим представлениям, жидкая вода содержит образованные водородными связями более или менее обширные псевдокристаллические группировки молекул Н2О. Такие постоянно разрушающиеся и вновь формирующиеся молекулярные агрегаты (“кластеры”) как бы плавают в моногидрольной воде (относительное количество которой может быть и небольшим). Нагревание способствует разрушению кластеров и смещению равновесия в пользу гидролей.
Химическойсистемой называется вещество или смесь веществ в определённом ограниченном объёме. Система может быть гомогенной и гетерогенной. Гомогенная система представляет собой единое по составу и внутренней структуре скопление частиц, либо одинаковых, либо разных, но полностью перемешенных друг с другом. Такой системой будет, например, вода, раствор сахара или соли, смесь газов, однородное твёрдое вещество и т. п. Наоборот, система является гетерогенной, если в ней одновременно содержатся различные по составу или внутренней структуре скопления частиц, отделённые друг от друга поверхностями раздела. К гетерогенным относятся, например, системы, состоящие из двух несмешивающихся жидкостей, льда и воды, смесей твёрдых веществ и т. д.
Ограниченные друг от друга составные части гетерогенной системы носят название её фаз. Фазой, таким образом, называется гомогенная часть гетерогенной системы. Например, система, состоящая из двух несмешивающихся жидкостей, двухфазна, смесь твёрдых веществ состоит из стольких фаз, сколько имеется этих веществ, и т. д. Очевидно, что всякая гомогенная система является вместе с тем и однофазной.
Сообщалось, что выдержанная при высокой температуре (под давлением) и затем охлаждённая вода по некоторым свойствам отклоняется от обычной, причём время её возвращения к “норме” составляет несколько суток. Если всё это верно, то данное сообщение весьма интересно.
Положение тройной точки на диаграмме состояния определяет типичный для данного вещества характер изменения агрегатных состояний при обычных условиях давления. Если точка эта лежит ниже 101325 Па, то последовательное нагревание твёрдого вещества переводит его сперва в жидкость и лишь затем в газ (пар). Напротив, если тройная точка лежит выше 101325 Па, то рассматриваемое вещество переходит из твёрдого состояния прямо в газообразное, т. е. при нагревании возгоняется. Из изложенного следует, что для получения возгоняющегося вещества в жидком состоянии нужно производить его нагревание под достаточно высоким давлением.
Международным соглашением (1954 г.) температура тройной точки на диаграмме состояния воды принята за основу абсолютной температурной шкалы с точным значением 273,16 К. Температура эта может быть экспериментально воспроизведена с точностью до 10-4 К.
Температурная шкала Цельсия, как и прежде, основывается на интервале между температурой плавления льда (0,0100 К ниже тройной точки) и температурой кипения воды под нормальным давлением.
Количество тепла, которое необходимо затратить для возгонки (сублимации) вещества, носит название его теплоты возгонки. Например для иода имеем I2(т) + 63 кДж = I2(г). Под температурой возгонки (т. возг.) понимается температура, при которой давление пара возгоняющегося вещества достигает 101325 Па.
Практически возгонка довольно часто наблюдается при обычном давлении и у веществ, тройная точка которых лежит ниже 760 мм рт. ст. Происходит это тогда, когда пару нагретого вещества обеспечен свободный уход из системы. Например, при нагревании достаточного количества твёрдого иода в колбе пары его вытесняют воздух и, находясь под давлением пара I2 в 101325 Па, иод плавится. Напротив, при нагревании в открытой чашке пары I2 не накапливаются и твёрдый иод испаряется, т. е. происходит его возгонка. Из изложенного следует, что то или иное поведение твёрдого вещества при нагревании определяется не общим внешним давлением, а создающимся в системе парциальным давлением его пара по отношению к задаваемому тройной точкой.
С химической точки зрения вода является весьма реакционноспособным веществом. Она соединяется со многими оксидами металлов и неметаллов, энергично взаимодействует с наиболее активными металлами и вступает в различные другие реакции самого разнообразного характера. Поэтому с проявлениями химических свойств воды придётся в дальнейшем встречаться довольно часто. При образовании рациональных названий химических соединений молекулы воды применяется термин “акво” или “гидрат” (в конце названия).
Роль воды в природе.
Вода покрывает около 3/4 всей земной поверхности. Общее её количество оценивается в 1,4·1018 т. Сосредоточена она главным образом в океанах и морях. Объём вод мирового океана составляет 1,37·109 км2 при средней глубине 3,8 км. В эпоху последнего большого оледенения Земли (около 15 тыс. лет тому назад) уровень мирового океана был примерно на 150 м ниже современного.
Из пресных вод земной поверхности основная доля (около 24 млн. км3) падает на ледяные массивы Антарктики (90%) и других континентов. Таяние всех льдов повысило бы уровень мирового океана на 56 м. Реки и озёра составляют вместе около 2 млн. км3. Атмосфера содержит около 14 тыс. км3 воды в виде пара. Если подытожить количество пресных вод земной поверхности, то получится приблизительно 26 млн. км3, т. е. количество, которое составляет около 2% от воды океана. Более или менее значительное содержание воды характерно для всех живых организмов. Например, тело человека (средняя плотность 1,07 г/см3) содержит её около 70 вес. %.
На протяжении известных нам геологических периодов количество свободной воды, по-видимому, сохранялось приблизительно постоянным. Хотя и в настоящее время действуют некоторые процессы, при которых она вступает в прочные соединения, однако проходят и обратные процессы, уравновешивающие эту потерю. В результате протекающих при высоких температурах и давлениях химических реакций между веществами глубинных слоёв земли образуются “ювенильные” воды (по приближённой оценки — 3·108 т ежегодно), которые затем выносятся на поверхность в виде водяного пара или горячих или холодных ключей. И те, и другие могут образоваться также за счёт обычных подпочвенных вод. Они часто содержат растворённые соли и газы. Тогда такие ключи называются минеральными источниками и частично используются для лечебных целей.
Большая теплоёмкость морской воде (в 33000 раз превышающая теплоёмкость равного объёма воздуха) определяет климатическую роль океанов. Мощные тёплые и холодные течения обуславливают климат омываемых ими частей суши. Например, климат Европы тесно связан с Гольфстримом, который гигантской струёй (25 млн. т нагретой до 26 °С воды в с) вытекает из Мексиканского залива, пересекает Атлантический океан, омывает берега Англии и Норвегии и теряется в Северном Полярном море. Конец его захватывает Кольский полуостров. Благодаря этому Мурманск является незамерзающей гаванью, Тогда как, расположенный значительно южнее Санкт-Петербургский порт зимой замерзает. Мягкость климата Западной Европы обусловлена именно влиянием Гольфстрима, в течение круглого года проносящего у её берегов большие массы нагретой воды, которая смягчает резкость температурных колебаний. В противоположность подобно “морскому” климату, “континентальный” климат удалённых от океана стран характеризуется резкой сменой температур по временам года. Вследствие той же причины — большей теплоёмкости воды — разница температур дня и ночи, очень резкая для стран с континентальным климатом, почти не заметна на островах океана.
Океан таит в себе огромные запасы энергии. Строго периодические приливы и отливы сопровождаются более или менее резкими изменениями уровня воды, доходящими на некоторых участках океанского побережья до 10 и даже 18 метром. Ориентировочно подсчитано, что общая мировая мощность приливной волны составляет 8000 млрд. кВт. В настоящее время ведётся проектирование и строительство ряда приливных гидроэлектростанций (ПЭС), а одна из них — на реке Ранс во Франции мощностью 240 тыс. кВт уже работает, давая ежегодно более 500 млн. кВт·ч. У нас работает опытная Кислогубская ПЭС (около Мурманска) и намечено проектирование Мезенской ПЭС мощностью в 1,5 млн. кВт с ежегодной выработкой 6 млрд. кВт·ч.
Растворяя газы атмосферы и перенеся их течениями на большие расстояния, океан, наряду с ветрами, выступает в роли регулятора состава воздуха. Особенно важна его роль для углекислого газа, которого океан содержит приблизительно в 25 (по другим данным — в 60) раз больше, чем атмосфера.
Путём испарения громадные количества воды постоянно переходят в атмосферу. Помимо прямого парообразования на свободной поверхности океана, рек и других водоёмов, большое значение имеет для этого процесса жизнедеятельность растений. Например, взрослая берёза извлекает корнями из почвы и испаряет с поверхности листьев до 700 л воды в сутки. Подобным же образом, за вегетационный период пшеница переводит из почвы в воздух около 2000 т воды с гектара.
Подсчитано, что по всему земному шару ежегодно испаряется около 520 тыс. км3 воды (причём на это затрачивается около 20% всей получаемой Землёй солнечной энергией). Зависимость средней влажности воздуха над океаном (в % водяного пара по объёму) от северной географической широты и времени года видна из таблицы:
| Широта | 0° | 10° | 20° | 30° | 40° | 50° | 60° | 70° | 80° |
| Влажность (%) | |||||||||
| Январь | 2,8 | 2,5 | 1,8 | 1,3 | 0,7 | 0,4 | 0,2 | 0,1 | 0,009 |
| Июль | 2,8 | 2,7 | 2,5 | 2,3 | 1,8 | 1,4 | 1,2 | 0,9 | 0,5 |
Содержащийся в воздухе водяной пар (наряду с углекислым газом) играет громадную роль в тепловом балансе земной поверхности: он пропускает бульшую часть солнечных лучей, но в значительной степени задерживает обратное тепловое излучение Земли и таким образом способствует сохранению ею тепла.
Попадая в верхние холодные слои воздуха, водяные пары сгущаются в мелкие капельки, которые образуют облака. Последние, перемещаясь вместе с воздушными течениями, уносят воду далеко от места первоначального испарения и в конце концов возвращают её Земле в виде амфотерных осадков (дождя и снега). Осадки эти, помимо самой воды, обычно содержат небольшие количества растворённых солей. Так, осадки на территории России, характеризуются следующим средним содержанием солевых ионов:
| Ион | Na+ | Ca2+ | Mg2+ | K+ | HCO3- | SO42- | Cl- | NO3- |
| Содержание мг/л | 5,1 | 4,8 | 1,7 | 0,2 | 18,2 | 9,2 | 5,5 | 1,7 |
Общее количество выпадающих ежегодно осадков соответствует покрывающему весь земной шар слою воды толщиной 1 м (тогда как конденсация всей единовременно содержащийся в атмосфере влаги дала бы только слой в 24 мм). Распределение осадков по земной поверхности весьма неравномерно. Так, в Черрапунджи (Индия) среднегодовое количество осадков превышает 10 м, а в Каире оно близко к нулю. Неравномерно и обычное для той или иной местности распределение осадков по месяцам года.
Именно воды дождей дают возможность развитию жизни почти на всей твёрдой земной поверхности. В отдельных засушливых областях из роль берут на себя воды рек (ежегодный мировой сток которых составляет около 38 тыс. км3). Пользуясь водой рек и применяя искусственное орошение, можно оживить громадные области пустынь, что и делается, например, в Средней Азии, где с помощью орошения могут быть использованы для получения громадных количеств электрической энергии. Общая учтённая энергетическая мощность рек России превышает 150 млн. кВт.
Падая на горные массивы, воды дождей частично задерживаются в их трещинах. Зимой, при замерзании воды, образующийся лёд расширяет эти трещины, раскалывает горные породы и постепенно превращает утёсы в груду обломков. Находясь под постоянным воздействием воды, воздуха и смены температур, эти обломки всё более раздробляются. Воды дождей извлекают из них растворимые составные части и вместе с захватываемыми в виде взвесей нерастворимыми частицами (главным образом песка и глины) уносят в реки. Здесь взвешенные частицы сортируются по удельному весу: сначала отлагается песок, дальше, в местах с более медленным течением, оседает глина. В течение веков вдоль русла реки образуются таким образом мощные залежи песка и глины, вследствие чего дно поднимается и сама река перемещается, прокладывая себе путь по новому направлению. На обнажившиеся старом русле начинает образовываться почва и развиваться наземная растительность.
Если в разрушаемой горной породе кроме песка и глины содержались какие-либо другие нерастворимые составные части, они также сортируются водой по их удельному весу. Так возникают залежи некоторых полезных ископаемых, например золота: его тяжёлые частички оседают вместе с более крупными зёрнами песка сравнительно близко к местам разрушения горных пород, образуя золотоносные россыпи.
Взвешенные в воде рек мельчайшие частицы (так называемый ил) иногда состоит из веществ, необходимых для питания растений. В таких случаях особенно большое значение имеют весенние разливы рек, так как при них часть ила оседает на почве окружающих равнин и увеличивает их плодородие. Хорошо известна в этом отношении, например, роль разливов Нила. Этим же отчасти обусловлен повышенный урожай трав на заливных лугах. Около 15 км3 осадков выносятся ежегодно реками всего мира в океан.
По пути к нему воды рек поглощают из воздуха значительное количество углекислого газа, способствующего (благодаря химическим реакциях) растворению минеральных пород по которым проходит русло реки. Поэтому по мере приближения к морю содержание растворённых веществ увеличивается. Насколько различно оно может быть в отдельных реках, показывают следующие примерные данные (мг/л):
Са2+ | Mg2+ | Na++K+ | HCO3- | SO42- | Cl- | |
| Нева | 8,0 | 1,2 | 3,8 | 27,5 | 4,5 | 3,8 |
| Эмба | 166 | 47 | 333 | 246 | 346 | 595 |
| Среднее по СНГ | 16,7 | 4,4 | 7,7 | 59,0 | 14,7 | 8,4 |
Из данных видно, что содержание солей в речных водах СНГ составляет немногим более 0,01 %. Примерно таковы же данные и по другим рекам земного шара. Несмотря на относительно малое содержание растворённых солей, их ежегодно выносится реками в океан более двух миллиардов тонн. Вычислено, например, что Волга ежегодно выносит в Каспийское море около 50 млн. т растворённых солей.
Содержание солей в морской воде несравненно больше, чем в речной. Для океана оно составляет в среднем 3,5 %, а у более или менее замкнутых морей колеблется в зависимости от многоводности впадающих рек и климатического пояса, в котором расположено мере. Так, солёность Средиземного моря доходит до 3,9 %, тогда как солёность Балтийского составляет в среднем лишь 0,5 %. Чёрное море содержит 1,8 % растворённых солей. Общее содержание солей мирового океана оценивается в 5·1016 т (из которых около 3/4 приходится на долю хлорида натрия). Среднее содержание важнейших ионов океанической воды (в процентах от их весовой суммы) таково:
Na+ | Mg2+ | Ca2+ | K+ | Cl- | SO42- | HCO3- | Br- |
| 30,6 | 3,7 | 1,2 | 1,1 | 55,2 | 7,7 | 0,3 | 0,2 |
Следовательно среди солей океана значительно преобладают хлористые и сернокислые соединения натрия и магния. По-видимому, основное количество входящих в состав солей океана неметаллов (Сl, S, Br и др) выделялось некогда вместе с парами самой воды из горячих недр Земли, тогда как основное количество металлов (Na, Mg, Ca и др) накапливалось в результате разрушения твёрдых горных пород земной поверхности.
Наименее растворимые составные части морской воды непрерывно оседают на дно океана. Вычислено, что ежегодно таким образом отлагаются около 2300 млн. т солей, из которых главная часть приходится на СаСО3. В результате образуются мощные залежи известняка и мела, который представляет собой скопление останков микроскопических раковин морских инфузорий, строивших свои жилища из растворённого в воде углекислого кальция. Эти залежи могут, однако, накапливаться лишь в сравнительно не глубоких местах океана, т. к. на больших глубинах, вследствие увеличения содержания растворённого углекислого газа, оседающий СаСО3 вновь растворяется. В глубоких местах дно океана покрыто особой красной глиной, образовавшейся, по-видимому, за счёт тепла вулканических извержений и космической пыли.
В результате сдвига земной коры моря на протяжении истории земли не раз меняли свои места. Например, ещё в сравнительно недавнюю геологическую эпоху южная половина СНГ представляла собой сплошное море, которое затем вошло в свои теперешние берега. Подобным же образом была в своё время покрыта водой Западная Сибирь. На поверхности поднявшегося морского дна оставались залежи известняка, мела и т. п. и ряд соляных озёр, частью уже высохших, частью же существующих до нашего времени (Каспийское и Аральское моря, озёра Эльтон, Баскунчак и др.). Высохшее морское дно в течение веков покрылось слоем почвы более или менее глубоко скрывшей под собой отложение некогда бывшего здесь моря.
Процесс усыхания замкнутых соляных озёр протекает и в настоящее время. Особенно интересный пример представляет залив Кака-Богаз-Гол на Каспийском море. С громадной поверхностью этого залива (18 тыс. км2) вода испаряется очень интенсивно, что вызывает постоянное поступление в него через узкий и мелкий пролив всё новых количеств морской воды. В результате этого протекающего тысячелетия процесса солёность Кара-Богаз-Гола во много раз превысила солёность самого Каспийского моря (в среднем 1,3 %). Вода Кара-Богаз-Гола содержит около 30 % растворённых солей и представляет собой настолько крепкий рассол, что при похолодании из неё выделяется кристаллический осадок (главным образом Na2SO4·10H2O), вновь растворяющийся при потеплении воды.
Богатым содержанием растворённых солей характеризуется ряд озёр Юговостока СНГ и Западной Сибири, Причём их солевой состав часто бывает различен. Встречаются озёра с преобладанием солей хлористых, сернокислых, углекислых, борнокислых, магнезиальных и т. д. Благодаря высокой концентрации растворённых солей такие озёра могут служить мощными сырьевыми базами для развития химических производств.
Однако несравненно более важна пресная вода. Для обеспечения физиологического равновесия человек должен ежедневно потреблять 2-4 литра воды (как таковой и с пищей). Фактически городской житель расходует на бытовые нужды в 100-200 раз больше (например, среднее для Москвы 560 л).
Помимо своего исключительного значения для жизни природы, вода является важнейшим и наиболее разносторонним по характеру объектом промышленного использования. Она применяется как исходное вещество, участник реакции или растворитель при проведении различных химических процессов, как теплоноситель и теплопередатчик в теплотехнике, как механическая сила при размыве грунтов и т. д. и т. п. Общее потребление воды для технических целей колоссально так, одна лишь металлургия расходует её больше, чем тратит на бытовые нужды всё население промышленно развитой страны.
В связи с этим для ряда стран всё возрастающее значение приобретает проблема пополнения своих природных водных ресурсов за счёт опреснения морской воды. Осуществляется оно различными методами (в основном — теми или иными вариантами перегонок), причём производительность уже действующих опреснительных установок достигает десятки тысяч кубометров за сутки.
Весьма перспективен, в частности, метод “мгновенной многоэтажной дистилляции”, основанный на понижении температуры кипения воды по мере уменьшения внешнего давления. Первоначально нагретая до 90 °С морская вода вводится в резервуар с пониженным давлением, где она бурно вскипает. Пар отводится и конденсируется, а вода переводится в следующий резервуар с более низким давлением, где процесс повторяется, и т. д.
Пероксид водорода
Кроме воды, известно другое соединение водорода с кислородом — пероксид водорода (Н2О2). В природе он образуется как побочный продукт при окислении многих веществ кислородом воздуха. Следы его постоянно содержатся в атмосферных осадках. Пероксид водорода частично образуется также в пламени горящего водорода, но при остывании продуктов сгорания разлагается.
В довольно больших концентрациях (до нескольких процентах) Н2О2 может быть получена взаимодействием водорода в момент выделения с молекулярным кислородом. Пероксид водорода частично образуется также при нагревании до 2000 °С влажного кислорода, при прохождении тихого электрического разряда сквозь влажную смесь водорода с кислородом и при действии на воду ультрафиолетовых лучей или озона.
Непосредственно определить теплоту образования пероксида водорода из элементов не удаётся. Возможность найти её косвенным путём даёт установленный Г. И. Гессом (1840 г.) закон постоянства сумм тепла: общий тепловой эффект ряда последовательных химических реакций равен тепловому эффекту любого другого ряда реакций с теми же самыми исходными веществами и конечными продуктами.
Строго говоря, закон Гесса следовало бы сформулировать, как “закон постоянства сумм энергий”, потому что при химических превращениях энергия может выделяться или поглощаться не только в тепловой, но и как механическая, электрическая и др. Кроме того, предполагается, что рассматриваемые процессы протекают при постоянном давлении или постоянном объёме. Как правило, именно так и обстоит дело при химических реакциях, а все другие формы энергии могут быть пересчитаны на тепловую.
Сущность этого закона особенно наглядно выявляется в свете следующей механической аналогии: общая работа, производимая опускающимся без трения грузом, зависит не от пути, а только от разности начальной и конечной высот. Подобным же образом общий тепловой эффект той или иной химической реакции определяется только разностью теплот образования (из элементов) её конечных продуктов и исходных веществ. Если всё эти величины известны, то для вычисления теплового эффекта реакции достаточно из суммы теплот образования конечных продуктов вычесть сумму теплот образования исходных веществ. Законом Гесса часто пользуются при вычислении теплот таких реакций, для которых прямое экспериментальное их определение трудно или даже невозможно.
В применении к Н2О2 Расчёт можно провести на основе рассмотрения двух различных путей образования воды:
1. Пусть первоначально при соединении водорода и кислорода образуется пероксид водорода, который затем разлагается на воду и кислород. Тогда будем иметь следующие два процесса:
2 Н2 + 2 О2 = 2 Н2О2 + 2х кДж
2 Н2О2 = 2 Н2О + О2 + 196 кДж
Тепловой эффект последней реакции легко определяется экспериментально. Складывая почленно оба уравнения и сокращая одиночные члены, получаем
2 Н2 + О2 = 2 Н2О + (2х + 196) кДж.
2. Пусть при соединении водорода с кислородом непосредственно образуется вода, тогда имеем
2 Н2 + О2 = 2 Н2О + 573 кДж.
Так как в обоих случаях и исходные вещества, и конечные продукты одинаковы, 2х + 196 = 573, откуда х = 188,5 кДж. Это и будет теплота образования моля пероксида водорода из элементов.
Пероксид водорода проще всего получать из пероксида бария (ВаО2), действуя на неё разбавленной серной кислотой:
ВаО2 + Н2SO4 = BaSO4 + H2O2.
При этом наряду с пероксидом водорода образуется нерастворимый в воде сульфат бария, от которого жидкость может быть отделена фильтрованием. Продаётся Н2О2 обычно в виде 3%-ного водного раствора.
Продолжительным упариванием обычного 3%-ного водного раствора Н2О2 при 60-70 °С можно довести содержание в нём пероксида водорода до 30%. Для получения более крепких растворов отгонку воды приходится производить под уменьшенным давлением. Так, при 15 мм рт. ст. сначала (примерно с 30 °С) отгоняется главным образом вода, а когда температура достигает 50 °С, в перегонной колбе остаётся очень концентрированный раствор пероксида водорода, из которого при сильном охлаждении могут быть выделены его белые кристаллы.
Основным методом получения пероксида водорода является взаимодействие с водой надсерной кислоты (или некоторых её солей), легко протекающее по схеме:
Н2S2O8 + 2 H2O = 2 H2SO4 + H2O2.
Меньшее значение имеют некоторые новые методы (разложение органических пероксидных соединений и др.) и старый способ получения из ВаО2. Для хранения и перевозки больших количеств пероксида водорода наиболее пригодны ёмкости из алюминия (не ниже 99,6%-ной чистоты).
Чистый пероксид водорода — бесцветная сиропообразная жидкость (с плотностью около 1,5 г/мл), под достаточно уменьшенным давлением перегоняющуюся без разложения. Замерзание Н2О2 сопровождается сжатием (в отличие от воды). Белые кристаллы пероксида водорода плавятся при -0,5 °С, т. е. почти при той же температуре, что и лёд.
Теплота плавления пероксида водорода составляет 13 кДж/моль, теплота испарения — 50 кДж/моль (при 25 °С). Под обычным давлением чистый Н2О2 кипит при 152 °С с сильным разложением (причём пары могут быть взрывоопасны). Для его критических температуры и давления теоретически рассчитаны значения 458 °С и 214 атм. Плотность чистого Н2О2 равна 1,71 г/см3 в твёрдом состоянии, 1,47 г/см3 при 0 °С и 1,44 г/см3 при 25 °С. Жидкий пероксид водорода, подобно воде, сильно ассоциирована. Показатель преломления Н2О2 (1,41), а также её вязкость и поверхностное натяжение несколько выше, чем у воды (при той же температуре).
Структурная формула пероксида водорода Н-О-О-Н показывает, что два атома кислорода непосредственно соединены друг с другом. Связь это непрочна и обусловливает неустойчивость молекулы. Действительно, чистая Н2О2 способна разлагаться на воду и кислород со взрывом. В разбавленных водных растворах она значительно устойчивее.
Оптическими методами установлено, что молекула Н-О-О-Н не линейна: связи Н-О образуют углы около 95° со связью О-О. Крайними пространственными формами молекул подобного типа являются показанные ниже плоские структуры — цис-форма (обе связи Н-О по одну сторону от связи О-О) и транс-форма (связи Н-О по разные стороны).
Н Н Н
| | Ѕ
О ѕ О О ѕ О
Ѕ
Н
Переход от одной из них к другой мог бы осуществляться путём поворота связи Н-О по оси связи О-О, но этому препятствует потенциальный барьер внутреннего вращения, обусловленный необходимостью промежуточного преодоления менее энергетически выгодных состояний (на 3,8 кДж/моль для транс-формы и на 15 кДж/моль для цис-формы). Практически круговое вращение связей Н-О в молекулах Н2О2 не осуществляется, а происходит только некоторые их колебания около наиболее устойчивого для данной молекулы промежуточного состояния — косой (“гош”) — формы.
Чем чище пероксид водорода, тем медленнее она разлагается при хранении. Особенно активными катализаторами разложения Н2О2 являются соединения некоторых металлов (Сu, Fe, Mn и др.), причём заметно действуют даже такие их следы, которые не поддаются прямому аналитическому определению. Для связывания этил металлов к пероксиду водорода в качестве “стабилизатора” часто добавляют немного (порядка 1:10 000) пирофосфата натрия — Na4P2O7.
Сама по себе щелочная Среда не вызывает разложения пероксида водорода, но сильно способствует её каталитическому распаду. Напротив, кислотная среда этот распад затрудняет. Поэтому раствор Н2О2 часто подкисляют серной или фосфорной кислотой. Разложение пероксида водорода идёт быстрее при нагревании и на свету, поэтому хранить его следует в тёмном прохладном месте.
Подобно воде, пероксид водорода хорошо растворяет многие соли. С водой (также со спиртом) она смешивается в любых соотношениях. Разбавленный его раствор имеет неприятный “металлический” вкус. При действии на кожу крепких растворов получаются ожоги, причём обожженное место окрашивается в белый цвет.
Ниже сопоставлена растворимость некоторых солей в воде и пероксиде водорода при 0 °С (г на 100 г растворителя):
| КСl | NaCl | NaNO3 | Na2SO4 | K2SO4 | |
Н2О | 28,2 | 35,6 | 73,3 | 4,9 | 7,3 |
Н2О2 | 63,3 | 20,5 | 30,9 | 26,7 | 96,1 |
Из приведённых примеров видно, что при переходе от Н2О к Н2О2 происходит не простое смещение растворимости в ту или иную сторону, а проявляется его сильная зависимость от химической природы солей.
Несмотря на большое сходство пероксида водорода с водой по составу и ряду свойств, смеси их замерзают при гораздо более низкой температуре, чем каждое вещество в отдельности. Существуют смеси замерзающие лишь ниже -50 °С. При таких условиях может образоваться очень нестойкое соединений состава Н2О2·2Н2О. Следует отметить, что содержащие более 50% Н2О2 водные растворы (равно как и безводный пероксид водорода) весьма склонны к переохлаждению. С эфиром пероксид водорода, подобно воде, смешивается лишь ограничено.
Пероксид водорода является сильным окислителем, т. е. легко отдаёт свой лишний (по сравнению с более устойчивым соединением — водой) атом кислорода. Так, при действии безводной и даже высококонцентрированной Н2О2 на бумагу, опилки и другие горючие вещества они воспламеняются. Практическое применение пероксида водорода основано главным образом на его окисляющем действии. Ежегодное мировое производство Н2О2 превышает 100 тыс. т.
Более половины всего вырабатываемого пероксида водорода расходуется на отбелку различных материалов, проводимую обычно в очень разбавленных (0,1-1%) водных растворов Н2О2. Важное преимущество пероксида водорода перед другими окислителями заключается в “мягкости” действия, благодаря чему сам отбеливаемый материал почти не затрагивается. С этим же связано и медицинское использование очень разбавленных раствором пероксида водорода в качестве антисептика (для полоскания горла и т. д.).
Очень концентрированные (80% и выше) водные растворы Н2О2 находят применение в качестве источников энергии и самостоятельно (с помощью катализаторов быстрого разложения Н2О2 из одного литра жидкого пероксида водорода можно получить около 5000 л нагретой до 700 °С смеси кислорода с водяным паром), и как окислитель реактивных топлив. Пероксид водорода применяется как окислитель в химических производствах, как исходное сырьё для получения пероксидных соединений, инициатор полимеризационных процессов, при изготовлении некоторых пористых изделий, для искусственного старения вин, крашения волос, вывода пятен и т. д.
Характерный для пероксида водорода окислительный распад может быть схематически изображён так:
Н2О2 = Н2О+ О (на окисление).
Кислая среда более благоприятствует этому распаду, чем щелочная.
Значительно менее характерен для пероксида водорода восстановительный распад по схеме:
Н2О2 = О2 + 2 Н (на восстановление)
Щелочная Среда более благоприятствует такому распаду, чем кислая.
Восстановительный распад пероксида водорода имеет место, например, в присутствии оксида серебра:
Ag2O + H2O2 = 2 Ag + H2O + O2.
Аналогично, по существу, протекает его взаимодействие с озоном (О3 + Н2О2 = 2 Н2О + 2 О2) и с перманганатом калия в кислой среде:
2 КMnO4 + 5 H2O2 + 3 H2SO4 = K2SO4 + 2 MnSO4 + 5 O2 + 8 H2O.
Последняя реакция применяется для количественного определения пероксида водорода.
Пероксид водорода обладает очень слабо выраженными кислотными свойствами. При её взаимодействии с гидроксидами некоторых металлов образуются соответствующие пероксиды, которые следует рассматривать как соли пероксида водорода. Так идёт реакция, например, с гидроксидом бария:
Ва(ОН)2 + Н2О2Ы ВаО2 + 2 Н2О.
Соли пероксида водорода характеризуются наличием в молекулах пероксидной цепочки из двух атомов кислорода. У нормальных оксидов подобные цепочки не имеется. Например:
Na-O-O-Na и О=С=О.
В связи с этим отношение пероксидов и нормальных оксидов к кислотам различно — первые реагируют с образованием пероксида водорода, а вторые дают воду:
ВаО2 + Н2SO4 = BaSO4 + H2O2
SnO2 + 2 H2SO4 = Sn(SO4)2 + 2 H2O
Путём изучения продуктов реакции с кислотами можно, таким образом, установить, является ли данное кислородное соединение пероксидом или оксидом.
Водородные атомы пероксида водорода могут быть замещены не только на металл, но и на некоторые радикалы кислотного характера. В последнем случае получаются кислоты, содержащие в составе молекулы пероксидную цепочку и называемые надкислотами. Они являются, следовательно, производными пероксида водорода (и подобно последней обладают сильными окислительными свойствами). Примером может служить надсерная кислота, схематическая формула которой: НO3S-O-O-SO3H.
Соли пероксида водорода являются наиболее обычными представителями пероксидов. Последние можно в общей формуле определить как химические соединения, содержащие непосредственно связанные друг с другом атомы кислорода. Обычные оксиды таких кислород-кислородных мостиков не содержат, чем принципиально и отличаются от пероксидов.
Сообщалось, что при взаимодействии Н2 и О2 с использованием электрического разряда удалось получить Н2О3. По данным инфракрасной спектроскопии, молекула имеет структуру О(ОН)2, причём связи О-О примерно на 5% длиннее и на 25% слабее, чем в Н2О2. При -60 °С разложение Н2О3 происходит за несколько часов на воду и кислород. В обычных условиях этот надпероксид совершенно неустойчив.
Алюминий
По распространённости в природе алюминий занимает четвёртое место (после О, Н, и Si), причём на его долю приходится около 5,5 % общего числа атомов земной коры. В своей геохимической истории алюминий тесно связан с кислородом и кремнием. Главная его масса сосредоточена в алюмосиликатах. Чрезвычайно распространённым продуктом разрушения образованных этими минералами горных пород является глина, основной состав которой (соответствующий каолину) отвечает формуле Al2O3·2SiO2·2H2O. Из природных форм нахождения алюминия наибольшее технологическое значение имеет боксит (Al2O3·xH2O) и криолит (AlF3·3NaF).
Первое выделение элементарного алюминия по схеме:
AlCl3 + 3 K = 3 KСl + Al
относится к 1825 г., но более или менее чистый образец получен лишь в 1827 г., когда и были впервые описаны свойства этого элемента. Технически его получали затем по реакции:
NaAlCl4 + 3 Na = 4 NaCl + Al,
и долгое время он ценился дороже золота. Природный алюминий состоит только из атомов 27Аl, т. е. является “чистым” элементом.
В основном состоянии атом алюминия имеет внешнюю электронную оболочку 3s23p1 и одновалентен. Возбуждение его до трёхвалентного состояния (3s13p2) требует затраты 347 кДж/моль.
Несмотря на наличие громадных количеств алюминия в почвах, растения, как правило, содержат мало этого элемента. Ещё значительно меньше его содержание в живых организмах. У человека оно составляет лишь десятитысячные доли процента по массе. Биологическая роль алюминия не выяснена. Явно выраженной токсичностью соединения его не обладают.
Элементарный алюминий получают электролизом раствора Al2O3 (“глинозёма”) в расплавленном криолите. Процесс ведут при температурах около 1000 °С в специальных электрических печах, причём на аноде выделяется кислород, а на катоде — жидкий алюминий. Последний собирается на дне печи, откуда его периодически и выпускают.
Очистка алюминия от примесей трудна, поэтому необходимо, чтобы чисты были сами исходные материалы для его получения. Криолит обычно готовят искусственно путём совместного растворения Al(OН)3 и соды в плавиковой кислоте по реакции:
3 Na2CO3 + 2 Al(OH)3 + 12 HF = 2 Na3AlF6 + 3 CO2 + 9 H2O.
Природные бокситы, в состав которых входит 50-60 % Al2O3 и ряд примесей (SiO2, Fe2O3 и др), подвергаются предварительной химической переработке с целью выделения из них достаточно чистого сесквиоксида алюминия (содержащей не более 0,2 % SiO2 и 0,04 % Fe2O3). Методы такой переработки сильно зависит от состава исходного боксита и довольно сложны.
В расплаве криолита (т. пл. 1012 °С) имеет место главным образом равновесие по суммарной схеме:
Na3AlF6Ы 3 Na+ + 2 F- + AlF4-.
Уже в гораздо меньшей степени протекает дальнейшая диссоциация тетрафторалюминат-иона:
AlF4Ы F- + AlF3Ы 2 F- + AlF2+Ы 3 F- + AlF2+Ы 4 F- + Al3+.
Растворённый в криолите глинозём диссоциирует по схеме:
Al2O3Ы AlO+ + AlO3-.
Так как между образующимися ионами возможны вторичные реакции (например, F- + AlO+Ы FAlO), ионно-молекулярный состав раствора Al2O3 в расплавленном криолите весьма сложен. Наинизшая температура плавления (665 °С) достигается в рассматриваемой системе при следующем её составе: 58 % криолита, 37 % AlF3 и 5 % Al2O3.
Печь для выплавки алюминия состоит из железного ящика, внутренние стенки и дно которого выложены теплоизолирующим слоем из огнеупорных материалов и поверх него — толстой угольной обкладкой, служащей при электролизе катодом. В качестве анода применяется массивный угольный электрод. Процесс ведут при температуре около 960 °С, напряжении около 5 В и силе тока около 140 тыс. А. Выделяющийся кислород образует с углём анода CO и CO2. Параллельно за счёт незначительного выделения фтора получаются небольшие количества CF4. Вследствие сгорания анода его приходится постепенно опускать вниз. Боковые стенки печи(и большая часть поверхности жидкости) покрыты твёрдой коркой электролита, препятствующий их разъединению выделяющимися у анода газами и предохраняющий расплав от охлаждения. Во время работы печи в неё периодически добавляется Al2O3 (и немного криолита), а расплавленный металл удаляется.
Выплавка алюминия весьма энергоемка: тонна металла требует затраты около 10 тыс. кВт·ч электроэнергии. Первичная его очистка осуществляется продувкой хлора. Продажный металл содержит обычно 99,7 % алюминия. Наряду с другими примесями (главным образом Si и Fe) в нём имеются и следы галия.
Очистка технического алюминия производится обычно при 710-740 °С электролизом в системе из трёх жидких фаз: катодом служит чистый алюминий (плотность 2,35 г/см3), электролитом — обладающая плотностью 2,7 расплавленная смесь солей (60 — BaCl2, 23 — AlF3, 17% — NaF), а анодом — исходный алюминий, к которому для повышения плотности добавляется до 25 % меди (что даёт плотность 3,3 г/см3). При этом трёхслойном электролизе металлы, располагающиеся в ряду напряжений правее алюминия (Cu, Ga и др.), в электролит не переходят, а располагающиеся левее — на катоде не выделяются. Таким путём алюминий доводится до чистоты более 99,99 %, а дальнейшая его очистка может быть в случае надобности проведена методом зонной плавки.
Алюминий представляет собой серебристо-белый, довольно твёрдый металл с плотностью 2,7 г/см3, плавящийся при 660 и кипящий при 2520 °С. Он характеризуется большой тягучестью и высокой электропроводностью, составляющей приблизительно 0,6 электропроводности меди. С этим связано его использование в производстве электрических проводов (которые при сечении, обеспечивающем равную электропроводность вдвое легче медных).
Наложением 6 тыс. атм давления порошок алюминия может быть превращён в компактный металл. Сжимаемость его сравнительно невелика (при 100 тыс. атм объём равен 0,92 атм от первоначального), а электросопротивление с повышением давления несколько уменьшается (составляя при 50 тыс. атм. около 0,8 от обычного). Плавление алюминия связано со снижением плотности от 2,55 до 2,38 г/см3. Теплота плавления равна 293, а теплота возгонки (при 25 °С) — 326 кДж/моль. В парах алюминий одноатомен.
Алюминий широко применяется для выделки домашней посуды, изготовления труб для нефтяной промышленности и дождевальных установок, сборных башен для хранения зерна. внешних обкладок электрических кабелей (вместо свинца) и т. д. Хотя алюминий примерно в 4 раза дороже железа, он начинает конкурировать с жестью в производстве консервных банок. Его высокая теплопроводность (почти в 3 раза превышающая теплопроводность железа) делает алюминий особенно пригодным для сооружения различных теплообменных установок. При 100-150 °С он настолько пластичен, что из него может быть получена фольга толщиной менее 0,01 мм. Подобная фольга применяется для изготовления электрических конденсаторов и для завертывания некоторых продуктов. Чистая алюминиевая поверхность отражает около 90 % падающего на неё излучения ( не только видимого, но также инфракрасного и ультрафиолетового). Поэтому нанесение на стекло алюминия (путём напыления в вакууме) позволяет получать высококачественные зеркала, очень равномерно отражающие лучи различных длин волн. Выдерживание тканей в высоком вакууме над жидким алюминием сопровождается их металлизацией (без потери проницаемости для воздуха). Помимо других применений, такие металлизированные ткани в сочетании с чёрными могут служить для регулирования температуры. Например, двухслойный плащ из них, надетый металлической стороной наружу (в жару), будет предохранять тело от перегревания, а надетый наружу чёрной стороной (в холод) — способствовать сохранению телом тепла. Тонкий порошок алюминия служит для изготовления устойчивой к атмосферным воздействиям серебряной краски, а также в качестве добавки к некоторым реактивным топливам. Алюминиевым остриём можно наносить чёткие надписи на стекло (предварительно обезжиренное и слегка влажное).
Пайка алюминевых изделий (после металлической зачистки поверхности может быть осуществлена при помощи сплава 60 % Sn + 40 % Zn или 60 % Zn + 40 % Cd. Хорошим флюсом служит при этом смесь из 8,6 % NH4BF4, 5,0 % — Cd(BF4)2 и 86,4 — триэтаноламина — N(CH2CH2OH)3 (масло с т. кип. 278 °С при 150 мм рт. ст.). Рекомендуется также предварительная протирка спаиваемых мест насыщенным раствором CuCl2 после чего пайка может проводиться обычным способом
Добавка к расплавленному алюминию подходящих вспенивающих веществ (например, MgH2) и разливкой в формы образовавшейся пены может быть получен пеноалюминий (“форалюм”). Он представляет собой микропористый металл с плотностью 0,4 г/см3, хорошо поддающийся обработке резанием. Детали из него можно скреплять даже гвоздями.
Алюминий может быть использован для выпрямления переменного тока. Выпрямитель составляется из малого (по поверхности) алюминиевого и относительно большого свинцового (или железного) электродов, погружённых в раствор буры (или 10 %-ный раствор (NH4)2CO3). Подобная система пропускает ток только в одном направлении — при котором Al является анодом — и выдерживает напряжение до 40 В. Для выпрямления тока более высокого напряжения алюминиевые выпрямители включают последовательно (а для получения достаточной силы тока — параллельно).
Важной областью использования Al является алитирование — насыщение поверхности изделий из стали или чугуна металлическим алюминием для придания им жаропрочности и предохранения от коррозии. Оно проводится обычно при 1000 °С в смеси, состоящей из порошкообразного алюминия (49), оксида алюминия (49) и хлористого аммония (2 %). Алитированные изделия можно нагреть до 1000 °С не опасаясь их окисления.
Значительно более обширно применение алюминия в виде различных сплавов, наряду с хорошими механическими качествами характеризующихся своей лёгкостью. Особенно важен так называемый дуралюмин (приблизительный состав: 94 % Al, 4 % Cu, по 0,5 % Mg, Mn, Fe и Si). Он ценен тем, что изделия из него при равной прочности в 3 раза легче стальных. Для авиационной промышленности лёгкость материала особенно важна, облегчение металлических конструкций имеет громадное значение для ряда областей техники. Это становится особенно наглядным, если принять во внимание, что, например, в гружёном товарном вагоне около трети всей массы приходится на метериалы, из которых изготовлен сам вагон, а в пассажирских вагонах на их собственную массу приходится до 95 % всей нагрузки. Очевидно, что даже частичная замена стали дуралюмином даёт громадный технико-экономический эффект. В связи с этим, а также ввиду наличия в природе практически неисчерпаемых запасом алюминия, его иногда называют “металлом будущего”. Возможность широкой замены им основного металла современной техники — железа — ограничивается главным образом сравнительно высокой стоимостью алюминия.
Помимо дуралюминия в технике используется и ряд других сплавов на основе алюминия. Из них следует отметить силумин (10-14% Si, 0,1 — Na), применяемый для изготовления различных частей, и гидроналий (3-12 % Mg), устойчивый к действию морской воды. Обладающие очень высокой коррозионной стойкостью сплавы алюминия, содержащие одновременно Mg и Si, являются основным материалом для изготовления несущих винтов вертолётов.
Мировая добыча алюминия достигала в 1885 г. лишь 13 т, в 1900 — 7 тыс. т, в 1935 г. — 260 тыс. т, в 1950 г. — 1,3 млн. т, а в настоящее время его ежегодная выработка составляет около 8 млн. т.
На воздухе алюминий покрывается тончайшей, но очень плотной плёнкой оксида, предохраняющей металл от дальнейшего окисления. В связи с этим поверхность его обычно имеет не блестящий, а матовый вид. При прокаливании мелко раздробленного алюминия он энергично сгорает на воздухе. Аналогично протекает и взаимодействие его с серой. С хлором и бромом соединение происходит уже при обычной температуре, с иодом — при нагревании. При очень высоких температурах алюминий непосредственно соединяется также с азотом и углеродом. Напротив, с водородом он не взаимодействует.
По отношению к воде алюминий практически вполне устойчив. Сильно разбавленные, а также очень крепкие HNO3 и H2SO4 на алюминий почти не действуют, тогда как при средних концентрациях этих кислот он постепенно растворяется. По отношении к CH3COOH и H3PO4 алюминий устойчив. Чистый металл довольно устойчив также и по отношению к соляной кислоте, но обычный технический в ней не растворяется. Алюминий легко растворим в сильных щелочах (NaOH, KOH) по реакции:
2 Al + 2 NaOH + 6 H2O = 3 H2 + 2 Na[Al(OH)4].
Довольно энергично разъедается он также раствором NH4OH. В ряду напряжений Al располагается между Mg и Zn. Во всех своих устойчивых соединениях он трёхвалентен.
Образующаяся на поверхности алюминия в атмосферных условиях плёнка оксида имеет обычно толщину менее 1 нм, но очень прочно связана с металлом. Искусственно получаемые действием окислителей плёнки значительно толще. Хорошая защитная плёнка может быть получена, например, погружением алюминия в раствор, содержащий 20 % Na2SO4 и 10 % HNO3. С помощью подобранных наполнителей таким плёнкам можно придавать различную окраску.
Напротив, после контакта алюминия с раствором HgCl2 плёнка эта становится столь рыхлой, что уже не защищает металл от дальнейшего окисления. В результате он быстро обрастает “бородой” из водного оксида (Al2O3·xH2O) и постепенно окисляется нацело. Получившийся водный оксид, и сам по себе и после обезвоживания нагреванием, обладает высокой сорбционной активностью.
При нагревании стойкость оксидной плёнки значительно снижается. Особо следует отметить возможность заметной растворимости алюминия при кипячении его с разбавленными растворами некоторых органических кислот.
Лёгкость растворения алюминия в сильных щелочах обусловлена снятием с него защитной оксидной плёнки по схеме:
Al2O3 + 2 OH’ + 3 H2O = 2 Al(OH)4’.
Так как в ряду напряжений Al стоит значительнее левее водорода, обнажение чистой поверхности металла тотчас сопровождается реакциями по схемам:
2 Al + 6H·(из воды) = 2 Al••• + 3 H2 и 2 Al••• + 8 OH’ = Al(OH)4’.
Равновесие первой из них всё время смещается вправо за счёт второй. Аналогично протекает растворение в щелочах и других активных металлов, гидроксиды которых амфотерны (Sn, Zn и т. п.). Переходу Al+3 + 3 e- = Al отвечают нормальные потенциалы -1,66 В (кислая среда) и -2,31 В (щелочная среда).
Соединение алюминия с кислородом сопровождается громадным выделением тепла, значительно большим, чем в случае многих других металлов. Ввиду этого при прокаливании смеси оксида такого металла с порошком алюминия происходит бурная реакция, ведущая к выделению из взятого оксида свободного металла. Метод восстановления при помощи Аl(алюмотермия) часто применяется для получения некоторых элементов (Cr, Mn, V и др.) в свободном состоянии.
Теплота образования Al2O3 из элементов составляет 1672 кДж/моль. Сжиганием порошка алюминия в токе кислорода может быть получено пламя с температурой до 3500 °С.
На этой основе был сконструирован “огненный нож”, образуемый пламенем взвешенной в кислороде смеси алюминиевого порошка с железным, вылетающим (под давлением) из длинной стальной трубы. При помощи такого “ножа” удавалось, в частности, разрезать бетонные блоки толщиной более трёх метров.
Лабораторное получение свободных элементов методом алюмотернии проводят обычно в шамотном тигле, который ставят на слой песка. Внутрь тигля закладывается смесь тонких порошков металлического алюминия и соответствующего оксида, а над ней горка смеси Al + BaO2, поджигаемая при помощи воткнутой в неё ленты металлического магния. Сверху всё засыпается порошкообразным СaF2, который в процессе реакции плавится и образует слой, изолирующий реакционную смесь от внешнего пространства.
Алюмотермией иногда пользуются для сварки отдельных стальных частей, в частности стыков трамвайных рельсов. Применяемая смесь (“термит”) состоит обычно из тонких порошков алюминия и железной окалины (Fe3O4). Поджигается она при помощи запала из смеси Al и BaO2. Основная реакция идёт по уравнению
8 Al + 3 Fe3O4 = 4 Al2O3 + 9 Fe + 3344 кДж,
причём развивается температура около 2500 °С. Помимо сварки, термит используется для переплавки стальных стружек (отходов металлообрабатывающей промышленности).
Сесквиоксид алюминия представляет собой белую очень тугоплавкую и нерастворимую в воде массу. Природный Al2O3 (минерал корунд), а также получаемый искусственно и затем сильно прокаленный, отличается большой твёрдостью и нерастворимостью в кислотах. В растворимое состояние сесквиоксид алюминия можно перевести сплавлением со щелочами или K2S2O7 по реакциям:
Al2O3 + 2 NaOH = H2O + 2 NaAlO2
Al2O3 + 3 K2S2O7 = Al2(SO4)3 + 3 K2SO4.
Обычно загрязнённый оксидом железа природный корунд вследствие своей чрезвычайной твёрдости применяется для изготовления шлифовальных кругов, брусков и т. п. В мелко раздробленном виде он под названием наждака служит для очистки металлических поверхностей и изготовления наждачной бумаги. Для тех же целей часто пользуются оксидом алюминия, получаемым сплавлением боксита (техническое название — алунд).
Чистый сесквиоксид алюминия (т. пл. 2050, т. кип. 3500 °С) непосредственно используется в производстве зубных цементов. Так, порошок одного из видов высококачественного зубного цемента получается сплавлением при 700-800 °С и последующим измельчением тщательно приготовленной смеси следующего состава: 28,4 % Al2O3,20,9-SiO2, 19,7-Na2SiF6, 19,0-CaSiF6, 3,9-CaCO3, 4,1-H3PO4, 4,0-H3AsO4. Жидкость для замешивания такого цемента представляет собой крепкий раствор Al(H2PO4)3.
Изделия из плавленного сесквиоксида алюминия имеют плотность 4,0 г/см3, обладают очень высокой механической прочностью и сохраняют её до 1800 °С. Исключительно велика и их химическая стойкость. Вместе с тем они хорошо проводят тепло и переносят температурные колебания. Напылением расплавленного сесквиоксида алюминия может быть создано эффективное защитное покрытие на металлах.
Сплавление равных по массе количеств Al2O3 и SiO2 с последующим выдуванием их расплава было получено стекловолокно (“файберфракс”), характеризующееся высокой термической устойчивостью и большой устойчивостью к химическим воздействиям. Оно не изменяет свои свойства до 1250 °С, плавится лишь выше 1600 °С и особенно пригодно для изготовления теплоизоляционных материалов.
На основе корунда был сконструирован сверхпрочный искусственный камень — “микролит”. Он состоит из очень мелких (порядка микронов) зёрен корунда с небольшой добавкой связывающего стеклообразного материала. Микролитовые резцы сохраняют свою чрезвычайную твёрдость до 1200 °С и допускают поэтому очень большую скорость металлообработки.
Прозрачные кристаллы корунда, красиво окрашенные незначительными примесями других веществ, известны в качестве драгоценных камней: красного рубина (окраска от примеси хрома), синего сапфира (следы Ti и Fe) и др. В настоящее время драгоценные камни на основе оксида алюминия (рубины, сапфиры и др.) делают искусственно путём сплавления и последующей кристаллизации Al2O3 в присутствии соответствующих примесей. Подобные искусственные камни по своим качествам лучше природных.
На кристалле рубина была впервые (1960 г.) реализована идей оптического квантового генератора (“лазера”) — устройства, создающего направленный пучок монохроматического (т. е. имеющего одну определенную длину волны) излучения в видимой области спектра или вблизи неё. Действие лазера (как и родственного ему “мазера”, генерирующего аналогичный пучок коротких радиоволн) основано на выделение энергии за счёт одновременно происходящего определённого снижения энергетического уровня множества одинаковых частиц.
Такими частицами в кристалле рубина являются примесные (порядка 0,05 вес. % Сr2O3) атомы трёхвалентного хрома. При действии на них света с длинами волн 610-380 нм происходит возбуждение этих атомов от основного до высоких энергетических уровней с которых электроны тотчас же — за время порядка стомиллионных долей секунды — самопроизвольно переходят на промежуточный уровень. Важной особенностью последнего является то, что он нём электроны способны удерживаться уже сравнительно долго — в течение тысячных долей секунды. Так как 10-8 сек « 10-3 сек, накопление электронов на этом уровне может оказаться очень значительным. Создающееся таким путём метастабильное состояние системы нарушается появлением введённого извне или возникшего в ней самой фотона с l = 694,3 нм, который индуцирует рабочий переход накопленных на этом уровне электронов, завершающийся за миллионные доли секунды.
Освещая специально подготовленный кристалл рубина достаточно мощными импульсами зелёного света, удаётся получить остро направленные пучки характерного для рубинового лазера красного света с длиной волны 694,3 пм.
Квантовые генераторы импульсного (как первый рубиновый) или непрерывного действия могут быть построены на основе рабочего вещества не только твёрдого, но и жидкого или газообразного. Например, весьма эффективен лазер, работающий на смеси Не + Ne и генерирующий красное излучение с l = 932,8 пм. Им широко пользуются, в частности, для снятия спектров комбинационного рассеяния.
Несколько особняком стоит углекислотный лазер, работающий на смеси СО2 с N2 и He. Генерируя отвечающее одному из атмосферных “окон” излучение с l = 10,6 мк, он превосходит все другие лазеры по абсолютной выходной мощности в непрерывном режиме (60 кВт и более). Сообщалось, что строятся лазеры мощностью 1 МВт.
Что касается создаваемых лазерами импульсных мощностей (на отдельные доли секунды), то они способны превышать миллионы кВт. Важно, что эта энергия концентрируется не только во времени, но и в пространстве: плотность её может достигать миллиардов кВт/см2, что уже сопоставимо с плотностями энергии, характерными для атомных ядер.
Очень перспективы лазеры на полупроводниках, так как они допускают непосредственное преобразование электрической энергии в световую и могут иметь очень высокий коэффициент полезного действия. Для возможности его работы важно, чтобы число электронов в зоне проводимости полупроводника n-типа и число дырок в валентной зоне полупроводника р-типа было достаточно велико (такие полупроводники называются “вырожденными”). Под действием постоянного тока высокого напряжения электроны и дырки движутся навстречу друг другу. Встречаясь в переходном слое (имеющем толщину порядка десятков микронов), они генерируют световые кванты.
Характеристики излучения отдельных квантовых генераторов весьма различны как по длинам генерируемых волн, так и по мощности лучевого пучка. Такой пучок может быть пригоден, например, и для глазных операций, и для прожигания отверстий в алмазах. Уже определилось множество областей возможного практического использования квантовых генераторов, и число их с каждым годом возрастает. В частности, монохроматический характер лазерного излучения при большой его мощности и уже частично освоенной методике главного измерения l открывает возможность избирательного стимулирования с помощью лазеров желаемых направлений химических процессов.
Ввиду нерастворимости Al2O3 в воде отвечающий этому оксиду гидроксид Al(OН)3 может быть получен лишь косвенным путём (исходя из солей). Он представляет собой объёмистый студенистый осадок белого цвета, практически нерастворимый в воде, но легко растворяющийся в кислотах и сильных щелочах. Гидроксид алюминия имеет, следовательно, амфотерный характер. Однако и основные, и особенно кислотные, его свойства выражены довольно слабо. В избытке NH4OH гидроксид алюминия нерастворим.
При взаимодействии Al(OH)3 с сильными щелочами образуются соответствующие алюминаты, например, по схеме:
NaOH + Al(OH)3 = Na[Al(OH)4].
Алюминаты наиболее активных одновалентных металлов в воде хорошо растворимы, но ввиду сильного гидролиза растворы их устойчивы лишь при наличии достаточного избытка щёлочи. Алюминаты, производящиеся от более слабых оснований, гидролизованы в растворе практически полностью и поэтому могут быть получены только сухим путём (сплавлением Al2O3 с оксидами соответствующих металлов). Большинство из них в воде нерастворимо.
Осаждение гидроксида алюминия в процессе нейтрализации кислого раствора происходит около рН = 4,5. Характер осадка существенно зависит от условий его образования. Продукт осаждения из кислых раствором аммиаком на холоду аморфен и содержит много воды, а осаждённый при нагревании (или достаточно долго стоящий под жидкостью) приблизительно отвечает составу Al2O3·H2O и при исследовании рентгеновскими лучами показывает наличие кристаллической структуры.
Микрокристаллическую структуру имеют и осадки состава Al(OH)3 получаемые из щелочных растворов (например, путём насыщения их СО2). При очень медленном выделении из щелочных растворов отдельные кристаллы Al(OH)3 достигают иногда такой величины, что становятся различны с помощью микроскопа. Кристаллические модификации гидроксида алюминия в форме минералов диспора (HAlO2), бемита [AlO(OH)] и гидраргаллита [Al(OH)3] составляет основу природных бокситов.
За исключением сред с pH > 13, где преобладают ионы AlO2’, щелочные растворы алюминатов содержат ионы [Al(OH)4]’, [Al(OH)5]”, [Al(OH)6]”’и различные полимерные анионы. При выделении из таких растворов некоторые алюминаты сохраняют состав гидроксидов (примером может служить Sr3[Al(OH)6]2), а другие подвергаются частичной дегидротации. Например, для кристаллического алюмината калия характерен состав 2KAlO2·3H2O, а не K[Al(OH)4] (т. е. KAlO2·2H2O).
Получаемые сплавлением Al2O3 с оксидами или карбонатами соответствующих металлов безводные алюминаты по своему составу производятся от HAlO2. Их образование иногда сопровождается значительным выделением тепла. Примером может служить реакция по уравнению:
Li2O + Al2O3 = 2 HAlO2 + 109 кДж.
Из относящихся сюда соединений следует специально отметить встречающуюся в природе обычную шпинель — Mg(AlO2)2 (т. пл. 2115 °С.
Несравненно большее, чем простые алюмосиликаты, распространены в природе различные алюмосиликаты, составляющие основную массу земной коры. Образование алюмосиликатов при её затвердевании протекало с поглощением тепла. В связи с этим выветривание их является процессом экзотермическим. Например, выветривание гранита сопровождается выделением 500 кДж на каждый килограмм минерала.
Главное направление химической стороны процесса выветривания горных пород заключается в выделении кремневых и алюмокремневых кислот угольной кислотой. Характер основных продуктов выветривания — SiO2 и каолина — различен. В то время как SiO2 представляет собой простейшее соединение кремния, каолин ввиду сложности его состава должен был бы рассматриваться скорее как промежуточное образование. Однако в главной своей массе он практически является конечным продуктом распада алюмосиликатов. Обусловлено это устойчивостью каолина по отношению к воде, воздуху,CO2 и нагреванию. Так содержащуюся в нём воду (точнее, её элементы) каолин отщепляет только около 500 °С.
Тем не менее некоторая доля природного каолина всё же подвергается дальнейшему разрушению. Однако оно обычно осуществляется лишь под воздействием живого вещества и, следовательно, представляет собой биохимический процесс. В результате его протекания кремний каолина переходит в SiO2·хН2О, а алюминий — в гидроксид или фосфат.
Чистый каолин представляет собой землистую белую массу нежную на ощупь. Обычно глины являются тесными смесями каолина с песком, известняком, оксидом железа и т. д., а также с ещё не успевшими выветриться частицами исходных минералов (полевых шпатов, слюд и др.). Глины с большим содержанием песка часто называют суглинками, а с большим содержанием СаСО3 (и MgCO3) — мергнлями. Окраска глин весьма разнообразна. Чаще всего встречаются бурые (от оксидов железа) или серые (от примеси органических веществ). Некоторые их сорта, интенсивно окрашенные оксидами Fe и Mn, используются в качестве минеральных красок (под техническим названиями: охра, умбра, сиенна и т. д.). Глины являются составной частью почв и часто образуют мощные пласты огромного протяжения.
Частицы каолина крайне мелкие и имеют пластинчатое строение, благодаря чему могут очень плотно соприкасаться друг с другом. Этим обусловлено важнейшее свойство глины — её водонепроницаемость. С этим же тесно связано другое весьма важное свойство глины — её пластичность, т. е. способность легко принимать и затем сохранять заданные формы. Большое значение для пластичности глин имеет то обстоятельство, что поверхность частиц каолина гидрофильна. Благодаря этому при замешивании с водой отдельные агрегаты частиц окружаются прочно адсорбированными на них водными оболочками, обеспечивающими скольжение таких агрегатов друг около друга. На высокой адсорбционной активности некоторых глин основано их техническое использование для обесцвечивания различных масел. Некоторые глины обладают также высокой каталитической активностью.
При соприкосновении с водой агрегаты частиц каолина заряжаются отрицательно. Добавление небольших количеств щёлочи вызывает сильное увеличение заряда за счёт дополнительной адсорбции ионов ОН’. В результате взаимного отталкивания частиц внутри агрегата последний распадается при этом на отдельные частицы, каждая из которых окружается собственной водной оболочкой. Процесс этот сопровождается дополнительным связыванием воды, и в присутствии небольших количеств щёлочи глина заметно “высыхает”. Так как, с другой стороны, частицы её сильно отталкиваются друг от друга, такая глина теряет пластичность и может быть насыпана в формы, что иногда бывает весьма важно. Сравнительно малая пластичность многих природных глин (в частности, самого каолина) обусловлена именно наличием в них небольших примесей щелочей. В подобных случаях эластичность может быть сильно повышена добавлением к глине небольших количеств какой-нибудь слабой кислоты, нейтрализующей избыточную щёлочь. Добавление к глине сравнительно больших количеств щёлочи вызывает, наоборот, разрядку отдельных частиц и агрегатов каолина и слипание их в ещё более крупные агрегаты. Так как в сумме на образование водной оболочки крупных агрегатов расходуется гораздо меньше воды, чем в случае мелких (а тем более — отдельных частиц), глина при этом заметно разжижается. Добавка достаточного количества щёлочи позволяет, следовательно, при замешивании глины обходиться значительно меньшим количеством воды, что иногда имеет большое значение.
Глина является основным сырьём керамической промышленности. Так называемая грубая керамика охватывает производство кирпича, различных огнеупорных (шамот и т. д.) и кислотоупорных материалов и изделий из глины, глиняной посуды (гончарное производство), изразцов, черепицы и т. д., а тонкая керамика — производство фарфора, фаянса и изделий из него. С технологической точки зрения глины делятся на “жирные” и “тощие”. Первые содержат сравнительно много каолина (и мало примесей). Они обычно обладают большой пластичностью и высокой огнеупорностью. Вторые, напротив, содержат много примесей. Как правило, они значительно менее пластичны и более легкоплавки.
Глины считаются огнеупорными, если они плавятся выше 1650 °С. Спекание начинается значительно ниже точки плавления (для чистого каолина — при 1400 °С, а для обычных глин — при более низких температурах). В результате полного спекания глиняной массы получается искусственный камень большой прочности, так называемый клинкер.
Употребляющийся выше шамот является самым распространённым огнеупорным материалом. Шамотный кирпич идёт на кладку печей, обмуровку паровых котлов и т. д. В состав шамотной массы обычно входит 50-65 % SiO2, 45-30 — Al2O3, 2 — CaO, 1,5 — MgO и 1,5 — Fe2O3.
Процесс керамического производства распадается обычно на следующие отдельные операции: 1, очистка глины (не всегда), 2) приготовление исходной смеси глины с песком, полевым шпатом и т. д. и замешивание её с водой, 3) формовка полученного теста, 4) сушка сформованного изделия, 5) его обжиг и 6) покрытие глазурью (не всегда). Очистка глины от примесей производится только в тех случаях, когда требуется большая чистота исходного материала (например, при производстве фарфора). Проводят её обычно путём отмучивания разболтанной с водой сырой глины: более тяжёлые частицы песка и т. п. быстро падают при этом на дно, а каолиновая взвесь переводится в отстойники, где и осаждается.
Состав исходной смеси сильно зависит от рода изделий. Кирпичная масса состоит обычно из смеси неочищенной тощей глины с большим количеством песка, фарфоровая или фаянсовая — из смеси каолина, кварца и полевого шпата и т. д. Фармовка изделий производится механически или же вручную на гончарных станках. Сушка сформованных изделий ведётся или просто на воздухе или в специальных сушилках. Температура обжига в зависимости от рода изделий обычно колеблется между 900 и 1400 °С.
В результате обжига получается твёрдый, но пористый предмет, который в случае надобности глазуруют. В состав глазури может входить ряд различных веществ: каолин, полевой шпат, кварц, борная кислота, оксиды металлов (обычно Pb и Sn) и т. д. После нанесения на обжигаемый предмет слоя глазури его подвергают вторичному обжигу при 1000-1400 °С. При этом глазурь сплавляется и образует стекло, закрывающее поры.
Обжиг керамических изделий наиболее экономично проводится в так называемых туннельных печах. Такая печь представляет собой длинный (50-150 м) узкий канал с нагревательным устройством в средней части. Через всю печь проходит рельсовый путь, по которому медленно движется состав из нагруженных обжигаемыми изделиями вагонеток. Необходимый для сгорания топлива воздух движется навстречу вагонеткам охлаждая уже обожженные изделия и нагревая ещё не поступившие в зону обжига. Благодаря этому достигается более полное использование тепла. Наряду с экономичностью в смысле расхода топлива туннельные печи характеризуются высокой производительностью, так как процесс обжига осуществляется в них непрерывно.
Керамическое производство является одним из самых старых в истории человечества. Кирпич вырабатывался в Египте ещё за 6000 лет до нашей эры. Там же в глубокой древности существовало гончарное производство.
Сплавлением каолина с содой и серой (или Na2SO4 и углам) получают важную минеральную краску — ультрамарин. В зависимости от условий получения он может быть различных цветов. Наибольшее практическое применение находит синий ультрамарин, служащий для изготовления масляной краски, окраски бумаги и т. д. Ввиду того, что его цвет хорошо нейтрализует жёлтые оттенки, обычный ультрамарин (“синька”) применяется для подсинивания белья, льна, крахмала и т. д. Состав его может быть приближенно выражен формулой Na7Al6Si6S2O24. Окраска обусловлена свободной серой или какими-либо сернистыми соединениями (возможно — S2-), коллоидно распределёнными в сплаве. По отношению к воздуху, воде и мылу ультрамарин устойчив, но кислоты (даже слабые) разлагают его с выделением сероводорода, элементарной серы и кремневой кислоты.
С кислотами Al(OH)3 образует соли, содержащие в растворе бесцветные ионы Al•••. Производные большинства сильных кислот хорошо растворимы в воде, но довольно значительно гидролизованы, и поэтому растворы их показывают кислую реакцию. Ещё сильнее гидролизованы растворимые соли Al3+ и слабых кислот. Многие из них (например, Al2S3) полностью разлагаются водой.
В водной среде ион Al3+ непосредственно окружён шестью молекулами воды. Такой гидратированный ион несколько диссоциирован по схеме:
[Al(OH2)6] Ы [Al(OH2)5OH]- + H+.
Константа его диссоциации равна 1·10-5, т. е. он является слабой кислотой (близкой по силе к уксусной). Октаэдрическое окружение Al3+ шестью молекулами воды сохраняется и в кристаллогидратах ряда солей алюминия.
В ряду бесцветных галогенидов алюминия AlF3 сильно отличается по свойствам от своих аналогов. Полученный сухим путём (например, прокаливанием Al2O3 в парах HF) фтористый алюминий представляет собой тугоплавкий кристаллический порошок. В воде он практически нерастворим.
Соединения алюминия с хлором, бромом и иодом легкоплавки, весьма реакционноспособны и хорошо растворяются не только в воде, но и во многих органических жидкостях. Взаимодействие безводных галогенидов с водой сопровождается значительным выделением тепла. В растворе все они сильно гидролизованы. Будучи заметно летучи уже при обычных условиях, AlCl3, AlBr3 и AlI3 дымят во влажном воздухе (вследствие гидролиза).
С галогенидными солями ряда одновалентных металлов галогениды алюминия образуют комплексные соединения, главным образом типов M3[AlF6] и M[AlГ4] (где Г — Cl, Br или I). Склонность к реакциям присоединения выражена у рассматриваемых соединений довольно сильно. Именно с этим связано важнейшее техническое применение AlCl3 — в качестве катализатора при переработке нефти и при органических синтезах.
Некоторые константы галогенидов алюминия сопоставлены ниже:
AlF3 | AlCl3 | AlBr3 | AlI3 | |
Плотность, г/см3 | 3,1 | 2,5 | 3,2 | 4,0 |
| Теплота образования кДж/моль | 1509 | 702 | 514 | 309 |
Температура плавления, °С | 1040 | 193 | 98 | 188 |
Температура кипения, °С | 1279 | 180 | 255 | 383 |
При нагревании AlCl3 возгоняется, и его температура плавления может быть определена только под давлением. Критическая температура AlCl3 равна 353 °С при критическом давлении 26 атм.
Интересным способом образования фтористого алюминия является нагревание Al2O3 до 450 °C в токе фтористого бора реакция идёт по уравнению:
Al2O3 + 3 BF3 = (OBF3) + 2 AlF3.
Безводный AlF3 практически нерастворим не только в воде, но и в жидком водороде.
Образующийся при взаимодействии Al(ОН)3 и НF водный фтористый алюминий малорастворим в воде и довольно сильно гидролизован. Из его раствора в водной НF выделяется обычно кристаллогидрат AlF3·3Н2О
Из продуктов присоединения к фтористому алюминию лучше других изучены его комплексные соли с фторидами одновалентных металлов типов M[AlF4], M2[AlF5] и главным образом M3[AlF6]. В виде кристаллогидратов Н3AlF6·6Н2О и Н3AlF6·3Н2О была выделена и отвечающая ему свободная гексафторалюминиевая кислота.
Ион [AlF4]-представляет собой тетраэдр [d(AlF) = 181 пм]. Последовательная диссоциация иона [AlF6]”’ в водном растворе характеризуется следующими константами (при 25 °С):
[AlF6]”’ | [AlF5]” | [AlF4]’ | AlF3 | AlF2• | AlF• | ||||
3·10-1 | 2·10-2 | 2·10-3 | 1·10-4 | 1·10-5 | 8·10-7 |
Полная константа диссоциации (константа нестойкости) этого иона равна их произведению, т. е. 1·10-20.
Для хлористого алюминия (но не для его аналогов) характерен своеобразный ход изменения электропроводности с температурой. По мере приближения к точке плавления электропроводность быстро возрастает, при переходе AlCl3 из твёрдого в жидкое состояние падает почти до нуля, а затем вновь начинает повышаться. При кристаллизации расплавленного AlCl3 наблюдается необыкновенно резкое уменьшение объёма (почти вдвое) и довольно значительное выделение тепла (35,5 кДж/моль). И то и другое обусловлено переходом хлористого алюминия от молекулярной структуры (в жидком состоянии) к ионной (в твёрдом). Кристаллизация AlBr3 и AlI3 сопровождается сравнительно малым уменьшением объёма (на 15 %) и гораздо меньшим выделением тепла (11,3 и 16,7 кДж/моль).
Плотности паров AlCl3, AlBr3 и AlI3 при сравнительно невысоких температурах более или менее точно отвечают удвоенным формулам — Al2Г6. При точках кипения диссоциированные части равны соответственно 0,02: 0,7 и 24 %. Димеризация по схеме: 2 AlГ3 ® Al2Г6 сопровождается довольно сильным выделением тепла: 122,2 (Cl), 110,8 (Br), 94,0 (I). Хлорид оказывается таким образом более склонным к димеризации, чем иод. Полностью он становится мономерным лишь выше 800 °С. Молекула AlCI3 плоская с d(AlCl) = 206 пм.
Рис. 1. Схема строения молекулы Al2Г6.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()

![]()
![]()
![]()

![]()
![]()
![]() Как видно из рис. 1, пространственная структура молекул Al2Г6 отвечает двум тетраэдрам с общим ребром. Каждый атом алюминия связан с четырьмя атомами галогена, а каждый из центральных атомов галогена — с обоими атомами алюминия. Из двух связей центрального галогена одна является донорно-акцепторной, причём алюминий функционирует в качестве акцептора. Обе связи неотличимы друг от друга. Структуры характеризуются следующими параметрами: d(AlГ(крайн)) = 204 (Cl), 222 (Br), 253 пм (I); РГAlГ(внешн) = 122° (Cl), 118° (Br), 112° (I); d(AlГ(средн)) = 224 (Cl), 238 (Br), 258 пм (I); РГAlГ(внутр) = 87° (Cl), 82° (Br), 102° (I). Для мономерной молекулы AlI3 было найдено значение d(AlI) = 244 пм. Аналогичную структуру имеет и Re2Cl6.
Как видно из рис. 1, пространственная структура молекул Al2Г6 отвечает двум тетраэдрам с общим ребром. Каждый атом алюминия связан с четырьмя атомами галогена, а каждый из центральных атомов галогена — с обоими атомами алюминия. Из двух связей центрального галогена одна является донорно-акцепторной, причём алюминий функционирует в качестве акцептора. Обе связи неотличимы друг от друга. Структуры характеризуются следующими параметрами: d(AlГ(крайн)) = 204 (Cl), 222 (Br), 253 пм (I); РГAlГ(внешн) = 122° (Cl), 118° (Br), 112° (I); d(AlГ(средн)) = 224 (Cl), 238 (Br), 258 пм (I); РГAlГ(внутр) = 87° (Cl), 82° (Br), 102° (I). Для мономерной молекулы AlI3 было найдено значение d(AlI) = 244 пм. Аналогичную структуру имеет и Re2Cl6.
Галогениды алюминия (кроме фторида) растворимы почти во всех органических растворителях, причём определение их молекулярных весов в таких растворах даёт различные результаты. Так, для AlBr3 в эфире и пиридине найден был простой молекулярный вес, а в CS2 — двойной. Обусловлено это различие тем, что пиридин и эфир значительно более полярны, чем CS2.
Безводный хлористый алюминий обычно получают либо нагреванием металлов в токе хлора или НCl, либо пропусканием хлора над нагретой до красного каления смесью Al2O3 c углём. Путём насыщения раствора Аl в соляной кислоте хлористым водородом может быть выделен бесцветный расплывающийся на воздухе кристаллогидрат AlCl3·6H2O со строением, отвечающим формуле [Al(OH2)6]Cl3 (при d(AlO) = 188 пм). Нагревание его ведёт к отщеплению воды с HCl с образованием в остатке оксида алюминия.
Растворимость хлористого алюминия в воде весьма велика и мало меняется с температурой — от 44 г при 0°С до 49 г AlCl3 на 100 г Н2О при 100 °С. Определяемая обычными экспериментальными методами степень гидролиза этой соли достигает в 0,1 н растворе 2 %. На самом деле происходит не только гидролиз, но и образование комплексных кислот типа H[AlCl3OH] и др.
С газообразным аммиаком хлористый алюминий образует бесцветный порошкообразный комплекс AlCl3·6NH3, частично отщепляющий NH3 лишь при 180 °С. Моноаммиакат AlCl3·NH3 плавится при 125 °С и перегоняется при 422 °С почти без разложения. Помимо аммиака хлористый алюминий способен присоединять оксиды азота, РН3, SO2, H2S, HCN и некоторые другие неорганические молекулы, а также многие органические вещества.
В эфирной среде были получены азид алюминия — Al(N3)3, его цианиды Al(CN)3 и Li[Al(CN)4]
Сернокислый алюминий бесцветен и легкорастворим в воде. Из растворов он выделяется обычно в виде кристаллогидрата Al2(SO4)3·18H2O. С сульфатами ряда одновалентных металлов сернокислый алюминий образует бесцветные комплексные соли типа M[Al(SO4)2]·12H2O. Будучи вполне устойчивы в твёрдом состоянии, эти соли (квасцы) в растворе сильно диссоциированы на отдельные составляющие их ионы. Помимо алюминия, комплексные сульфаты типа квасцов известны и для ряда других трёхвалентных металлов (Cr, Fe, V и др.). В качестве одновалентных катионов (M) в их состав могут входить K+, Na+, NH4+ и некоторые другие.
Растворимость сульфата алюминия в воде составляет (г сульфата алюминия на 100 г воды):
Температура, °С | 10 | 20 | 30 | 50 | 100 | |
| Растворимость | 31,2 | 33,5 | 36,2 | 40,5 | 52,2 | 89,1 |
Сульфат алюминия применяется, в частности, для очистки воды. При добавлении к последней Al2(SO4)3 и небольшого количества извести первоначально получаемый коллоидный раствор Al(OH)3, который затем коагулирует, давая объёмистый студенистый осадок, захватывающий в процессе своего образования взвешенные в воде частицы и бактерии и увлекающий их затем на дно отстойника. Так как осветление воды идёт при этом быстро, размеры отстойника могут быть небольшими, что практически весьма важно. При очистке жёсткой воды добавление извести часто оказывается излишним. Термическое разложение Al2(SO4)3 начинается при 530 °С и заканчивается (образованием Al2O3) при 860 °С. Для константы диссоциации иона AlSO4+• даётся значение К = 2·10-4.
Квасцы обычно растворимы значительно хуже, чем отдельные составляющие их сульфаты. При повышении температуры растворимость в большинстве случаев увеличивается очень сильно, как это видно, например, из приводимых ниже данных для алюмокалиевых квасцов (г KAl(SO4)2 на 100 г воды):
Температура, °С | 15 | 30 | 60 | 100 | |
| Растворимость | 3,0 | 5,0 | 8,4 | 24,8 | 154 |
В их 0,1 н. растворе рН = 3,2.
Алюмокалиевые квасцы KAl(SO4)2·12Н2О являются важнейшим представителем соединений этого типа. При крашении тканей они [и Al2(SO4)3] используются в качестве протравы, в кожевенной промышленности для “белого” дубления кож, в бумажной — при проклеивании бумаги. Последняя операция особенно необходима для писчих сортов, так как из непроклеенной (например, фильтровальной или газетной) бумаге чернила расплываются. В медицине квасцы используются как наружное вяжущее средство (например, для остановки кровотечения при мелких порезах). При 93 °С они плавятся в своей кристаллизационной воде и затем легко обезвоживаются. “Жжёные” (т. е. обезвоженные нагреванием) квасцы применяются иногда как средство от потения ног.
Из остальных производных алюминия следует упомянуть его ацетат Al(CH3COO)3, используемый при крашении тканей (в качестве протравы). Соль эту обычно получают (из Al(OH)3 и CH3COOH) прямо в растворе, где она очень сильно гидролизована. Нитрат алюминия легкорастворим в воде. Фосфат нерастворим в воде (и уксусной кислоте), но растворим в сильных кислотах и щелочах.
Ацетат алюминия был получен взаимодействием AlCl3 с уксусным ангидридом (СН3СО)2О при 180 °С. В индивидуальном состоянии соль эта может существовать лишь при полном отсутствии воды. Её водный раствор не только сильно гидролизован и легко выделяет основные соли, но также обладает малой электропроводностью, что указывает на наличие комплексообразования. Несравненно отчетливее выражено такое комплексообразование у ацетилацетоната алюминия — Al(C5H7O2)3 (т. пл. 192, т. кип. 315 °С). Средняя энергия его связи с кислородом оценивается в 268 кДж/моль.
Для нитрата алюминия характерен кристаллогидрат Al(NO3)3·9Н2О. Его взаимодействием с N2O5 может быть получен аддукт Al(NO3)3·N2O5, при последующей возгонке дающий безводный Al(NO3)3. От последнего производятся устойчивые лишь в отсутствие влаги комплексные соли типа M[Al(NO3)4]. Растворимость нитрата алюминия велика от 56 г при 0 °С до 120 г Al(NO3)3 на 100 г Н2О при 80 °С. Ещё более растворим его перхлорат — 1220 г при 0°С и 180 г Al(ClO4)3 на 100 г H2O при 90 °С.
Нормальный фосфат алюминия AlPO4 интересен тем, что точно повторяет диоксид кремния по кристаллическим модификациям. Обусловлено это одинаковостью сумм зарядов Al3+P3+и Si4+Si4+ при близких радиусах всех трёх элементов.
Сульфид алюминия Al2S3 может быть получен непосредственно из элементов. Реакция начинается при нагревании и сопровождается большим выделением тепла (723 кДж/моль). После очистки возгонкой Al2S3 представляет собой белые иглы (т. пл. 1120 °С). Расплавленный Al2S3 хорошо растворяет аморфный оксид алюминия и при охлаждении вновь выделяет его, но уже в кристаллическом виде. При прокаливании на воздухе Al2S3 сгоряет до Al2О3 и SО2. Водой он полностью разлагается на Al(OH)3 и Н2S, что может быть использовано для получения чистого сероводорода.
Нагреванием Al2S3 с избытком AlСl3 в запаянной трубке был получен тиохлорид алюминия — SAlCl. На воздухе это белое твёрдое вещество постепенно переходит в ОAlCl. Известен и жёлтый селенид алюминия Al2Se3, образующийся из элементов со значительным выделением тепла (543 кДж/моль). Его взаимодействием с водой удобно пользоваться для получения селеноводорода. Теллурид алюминия во влажном воздухе выделяет теллуроводород.
С азотом порошкообразный алюминий соединяется выше 800 °С, причём реакция сопровождается довольно значительным выделением тепла (318 кДж/моль AlN). Нитрид алюминия представляет собой белый порошок, не изменяющийся при нагревании до 1800 °С, а выше этой температуры начинающийся распадаться на элементы. Под давлением азота в 4 атм AlN плавится при 2200 °С. Получать его удобно нагреванием (NH4)3AlF6 до 500 °С в токе аммиака. Химическая стойкость нитрида алюминия сильно зависит от компактности образца. В виде рыхлого порошка он медленно разлагается водой по схеме:
AlN + 3 Н2О = Al(ОН)3 + NН3.
Напротив, кристаллический AlN практически не поддаётся действию кипящих сильных кислот и лишь медленно разлагается горячими растворами щелочей. Кристаллы его обладают высокой твёрдостью и довольно хорошей теплопроводностью, а по электрическим свойствам приближается к полупроводникам. Интересно, что нагревание алюминия выше 1750 °С в атмосфере N2 с примесью СО приводит к образованию не белого, а голубого AlN. Присутствие следов кремния сообщает AlN способность к фосфоресценции. При нагревании смеси Li3N и AlN образуется сероватый двойной нитрид Li3AlN2.
В техническом масштабе нитрид алюминия может быть получен прокаливанием до 1700 °С смеси оксида алюминия и угля в атмосфере азота по реакции:
Al2O3 + 3 C + N2 + 702 кДж = 3 CО + 2 AlN.
При пропускании над AlN перегретого водяного пара выделяется аммиак, а оксид алюминия регенерируется. На этом был основан один из предложенных, но не применявшихся в практике способов синтеза аммиака.
Серый фосфид AlP может быть получен взаимодействием элементов около 500 °С. Образуется он с выделением тепла (121 кДж/моль) и сам по себе устойчив по крайней мере до 1000 °С, но во влажном воздухе медленно разлагается на Al(ОН)3 и PН3. С этим связано его использование в зернохранилищах для обеззараживания зерна. Он обладает полупроводниковыми свойствами.
Практически важнее своих аналогов антимонид алюминия AlSb, так как он является полупроводником, близким по характеристике к Si и Ge, но более дешёвым. Образование этого соединения из элементов протекает с выделением тепла (96 кДж/моль). В сухом воздухе AlSb (т. пл. 1070 °С) устойчив, но во влажном постепенно разрушается.
Жёлтый карбид алюминия Al4C3 образуется при нагревании смеси оксида алюминия и угля приблизительно до 2000 °С. Реакция его образования из элементов протекает с выделением тепла (196 кДж/моль). Он растворим в жидком алюминии и может быть из него перекристаллизован. Выше 2000 °С карбид алюминия начинает испаряться с частичным разложением (общее давление достигает 1 атм при 2400 °С). Водой он разлагается по уравнению:
Al4C3 + 12 Н2О = 4 Al(ОН)3 + 3 СН4.
Нагреванием металлического алюминия в токе ацетилена до 500 °С может быть получен ацетиленид состава Al2(C2)3. Водой он разлагается с выделением ацетилена.
Сплавлением Al4C3 с Al2О3 были получены оксокарбиды алюминия — Al2OC (т. пл. 2200 °С) и Аl4O4C (т. пл. 1890 °С). Оба они постепенно разлагаются на воздухе.
Бориды алюминия имеют состав AlB2 и AlB12. Последний образуется в виде прозрачных, сильно преломляющих свет и очень твёрдых кристаллов при перекристаллизации бора из расплавленного алюминия. Он известен в двух формах, структуры которых пока точно не установлены. Оба борида водой не разлагаются и довольно устойчивы по отношению к кислотам.
Взаимодействие LiH с AlСI3 в тщательно обезвоженном эфире по реакции:
4 LiH + AlСI3 = 3 LiСI + LiAlН4
и последующим испарением жидкости (отфильтрованной от нерастворимых в эфире LiСI и LiH) может быть получен алюмогидрид (аланат) лития — LiAlH4. Он представляет собой бесцветное кристаллическое вещество, устойчивое ниже 125 °С, но под действием воды бурно разлагающееся с выделением водорода. Алюмогидрид лития хорошо растворим в эфире (21,3 при 0 °С и 28,3 масс. % при 25 °С) и характеризуется ещё более сильно выраженными восстановительными свойствами, чем соответствующий боргидрид (например, СО2 восстанавливается им до СН3ОН). Оба обстоятельства дают возможность проводить при его помощи разнообразные реакции восстановления органических соединений. Взаимодействие LiAlН4 с хлоридами ЭСl4 (где Э — Si, Ge, Sn) является удобным методом получения SiH4, GeH4 и SnH4.
Лежащий в основе аланатов тетраэдрический ион [AlН4]- имеет d(AlH) = 155 пм. Отвечающие ему соли натрия и калия могут быть получены реакциями обменного разложения по схемам:
NaH + LiAlH4 = LiH + NaAlH4 и KH + NaAlH4 = NaH + KAlH4,
а также прямым синтезом из элементов (в гептане при 150 °С под давлением водорода около 300 атм). По свойствам они похожи на LiAlH4, но нерастворимы в эфире и более устойчивы. Так, NaAlH4 плавится при 178 °С почти без разложения. Хорошим растворителем этих аланатов является тетрагидрофуран.
В эфирном растворе по схеме:
3 LiAlH4 + AlCl3 = 3 LiCl + 4 AlH3
образуется гидрид алюминия, постепенно выделяющийся в виде белой аморфной массы. Последняя представляет собой полимеризованный продукт основного состава (AlH3)n, но содержит также связанный эфир, от которого не может быть полностью освобождена без потери части водорода. Строение кристаллического (AlH3)n соответствует трёхмерной сетке, образованной мостиковыми связями Al···H···Al c d(AlH) = 172 пм. Для энергии связи AlH в мономерной молекуле даётся значение 330 кДж/моль. Выше 100 °С алюмогидрид разлагается на элементы, а водой быстро гидролизуется.
Аммиак действует на AlH3 иначе, чем триметиламин. При -80 °С в тетрагидрофуране образуется белый объёмистый осадок H3NAlH3, который уже около -30 °С с отщеплением водорода переходит в H2NAlH2, при комнатной температуре — в HNAlH и наконец около 150 °С — в AlN.
При взаимодействии гидрида алюминия с дибораном образуется Al(ВH4)3. Боранат является самым летучим соединением алюминия — давление его пара равно 120 мм рт. ст. уже при 0 °С. Он может быть получен по схеме:
3 NaBH4 + AlCl3 = 3 NaCl + Al(BH4)3
и на воздухе самовоспламеняется. Боронат алюминия считается перспективным горючим реактивных топлив. Для него известны ряд продуктов присоединения.
Выше примерно 900 °С становится более или менее устойчивым одновалентное производное алюминия. Отвечающее ему соединение частично образуется при высоких температурах по обратимым реакциям типа:
AlГ3 + 2 Al Ы 3 AlГ или Al2Э3 + 4 Al Ы 3 Al2Э
(где Э — О, S, Se), но не способны к существованию при низких.
Значительно лучше других соединений одновалентного алюминия изучен AlF, образование которого около 1000 °С описывается уравнением:
AlF3(т) + 2 Al(ж) + 226 кДж = 3 AlF(г).
Смещение последнего равновесия при изменении температуры может быть использовано для очистки алюминия: высокотемпературное взаимодействие AlF3 с исходным металлом даёт AlF, который в холодной зоне распадается выделяя более чистый металл. Процесс этот является типичной транспортной реакцией. Несколькими его повторениями удалось получить алюминий с чистотой в семь девяток. Контроль чистоты осуществляется определением электрического сопротивления образца (при температуре жидкого гелия), линейно зависящего от содержания примесей.
29
Атом.
Основные представления о внутреннем строении вещества
Реальность атомов и молекул.
Согласно кинетической теории газов лишь очень небольшая (при обычных условиях примерно одна десятитысячная) доля всего объёма газа занята самими молекулами, которые находятся в состоянии непрерывного беспорядочного движения. Каждая молекула ежесекундно несколько миллиардов раз сталкивается с другими, поэтому средняя длина её свободного пробега измеряется лишь десятками миллиметров.
Кинетические представления М.В. Ломоносова наиболее полно развиты в его работе “Опыт теории упругости воздуха” (1748 г.). “Атомы воздуха,— писал Ломоносов, — в нечувствительные промежутки времени сталкиваются с другими, сходными, в беспорядочной взаимности, и когда одни находятся в соприкосновении, другие отрываются друг от друга и снова сталкиваются с другими, более близкими, снова отскакивают, так что стремятся рассеяться во все стороны, постепенно отталкиваемые друг от друга такими очень частыми взаимными ударами”
Средняя скорость молекул основных газов воздуха — азота и кислорода — составляет при обычных условиях около 460 м/с, среднее число столкновений каждой молекулы за секунду — около 7 миллиардов, а средняя длина свободного пробега — около 70 нм. Так как средняя длина свободного пробега обратно пропорциональна давлению газа, под вакуумом, например, в миллионную долю миллиметра ртутного столба она составляет уже около 50 м. Практически это означает, что молекулы при таком вакууме несравненно чаще будут сталкиваться со стенками заключающего газ сосуда, чем друг с другом.
Ударяясь о ту или иную преграду, молекулы производят на неё давление, которое является суммарным результатом толчков молекул. Оказываемое давление будет тем значительнее, чем больше толчков за единицу времени и чем сильнее каждый из них. Одним из важнейших выводов кинетической теории было то, что при данной температуре средняя кинетическая энергия поступательного движения молекул не зависит от их природы; иначе говоря, с извинением массы молекул скорости их изменяются так, что средняя кинетическая энергия остаётся постоянной. Поэтому давление должно зависеть только от числа молекул (в единице объёма).
В воздухе у земной поверхности площадь размером в 1 см3 испытывает 1023 ударов молекул за секунду. Но по мере удаления от земной поверхности давление воздуха уменьшается. Поэтому чем выше находится слой газа, тем меньше в нём концентрация молекул. Кинетическая теория даёт возможность рассчитывать изменение концентрации с высотой для частиц любой массы.
Очевидно, что если бы удалось доказать правильность расчётов кинетической теории при опытах с учётом поведения каждой отдельной частицы, то тем самым были бы подтверждены молекулярно-атомистические представления. Но главная сложность связана с ничтожными размерами молекул.
Перрен устранил это затруднение, воспользовавшись более крупными частицами. В результате долгой кропотливой работы ему удалось наделать из некоторых смолистых веществ шариков приблизительно одинакового радиуса — порядка десятых долей микрона. Такие частицы хорошо видны под микроскопом. Зная их радиус и плотность применённого для изготовления вещества, легко вычислить массу каждого шарика. Будучи разболтаны с водой (или другой жидкостью) в маленькой стеклянной камере, они первоначально занимают весь объём равномерно, но затем, после отстаивания, устанавливается определённое распределение частиц по высоте. Производя при помощи микроскопа подсчёт числа частиц в единице объёма на разных высотах можно проверить совпадают ли результаты с требованием кинетической теории.
Наиболее трудной частью исследования Перрена было приготовление шариков определённых размеров. “Мне пришлось,— пишет он,— обработать 1 кг гуммигута, чтобы получить через несколько месяцев фракцию, содержащую несколько дециграммов зёрен, диаметр которых был весьма близок к той величине, какую хотелось получить”. Сами опыты проводились при очень различных условиях: температура изменялась от –9 до +58 °С, вязкость среды — в отношении 1:330, масса шариков — в отношении 1:70 000 и т.д.
Подсчёт частиц на различных высотах производился в очень узком поле зрения, причём выводилось среднее из многих отдельных отсчётов. Например, при одном из опытов с гуммигутовыми шариками радиусом 0,21 мк отсчёты производились на высоте 5, 35, 65 и 95 мк от дна камеры. По теории, отношение числа частиц на этих высотах ожидалось в данном случае равным 100:48:23:11. При проведении опытов было пересчитано 13 000 шариков, причём результаты относительного распределения по высотам выразились цифрами 100:47:23:12.
Совпадение результатов Перрена с требованиями кинетической теории как при распределении частиц по высоте, так и при проверке других вытекающих из этой теории следствий получилось блестяще. После этого стало уже невозможно возражать против реальности молекул, и приблизительно к 1910 г. молекулярно-атомистические представления вновь стали общепринятыми.
Еще значительно раньше, во второй половине ХIХ века, были сделаны попытки подойти к вопросу об абсолютной массе и размерах атомов и молекул. Взвесить отдельную молекулу явно невозможно, однако теория открыла другой путь: надо было как-то определить число молекул в моле — так называемое число Авогадро (N). Непосредственно сосчитать молекулы так же невозможно, как и взвесить их, но число Авогадро входит во многие уравнения различных отделов физики, и его можно, исходя из этих уравнений, вычислить. Очевидно, что если результаты таких вычислений, произведённых несколькими независимыми путями, совпадут, то это может послужить доказательством правильности найденной величины.
Результаты первых определений числа Авогадро
Метод N.1023 Метод N.1023
Голубой цвет неба 6,04 Радиоактивные явления 6,04
Теория излучения 6,05 Структура спектральных линий 6,08
Распределение частиц по высоте 6,05 Строение кристаллов 6,04
Электрические заряды частиц 6,02 Поверхностное натяжение растворов 6,00
Результаты первых определений числа Авогадро сопоставлены выше. Все они, несмотря на различие использованных методов, очень близки друг к другу. В настоящее время значение числа Авогадро принимается равным 6,02.1023. Некоторое представление о громадности этой величины можно получить, исходя из следующих данных: если бы всё население Земли (около 4 миллиардов человек) стало бы считать молекулы, содержащиеся в одном моле, то при непрерывном отсчёте каждым человеком по одной молекуле в секунду для выполнения работы потребовалось бы около 5 миллионов лет.
Уточнённое значение числа Авогадро равно (6,0225±0,0003).1023. На его основе формулируется расширенное понятие моль; как число единиц любого вида (молекул, атомов, электронов и др.), равное числу Авогадро.
Зная число Авогадро, легко найти абсолютную массу частицы любого вещества. Действительно, абсолютная масса (в граммах) единицы атомных и молекулярных весов равна 1/N, т. е. 1,66.10–24 г. Масса эта во столько же раз меньше массы маленькой дробинки, во сколько раз масса человека меньше массы всего земного шара.
Пользуясь числом Авогадро, можно оценить также размеры атомов. Например, атомный вес натрия равен 23,0 и плотность его — 0,97 г/см3. Объём, занимаемый молем натрия (т. н. атомный объём), равен, следовательно, 23: 0,97 = = 23,7 см3. Так как моль содержит 6,02.1023 атомов, на долю каждого приходится 23,7/6,02.1023 = 3,9.10–23 см3, что соответствует кубику с длиной ребра 340 пм.
В действительности правильнее рассматривать атомы не как кубики, а как шары, причём определение радиуса атома Na более точными методами даёт 186 пм. Радиусы других атомов также выражаются величинами порядка сотен пикометров.
Сложность структуры атома.
До конца прошлого столетия физика и химия имели мало точек соприкосновения. Лишь в ХХ веке была стёрта резкая граница между обеими науками. Пограничные области назывались физической химией и химической физикой.
Хотя начало первой из них было положено ещё М.В. Ломоносовым (1752 г.), широко развилась она лишь в конце ХIX века и имела своим содержанием применение к обычным химическим проблемам теоретических и экспериментальных методов физики. Областью второй, целиком развившейся в ХХ веке, являлось изучение внутреннего строения атомов и молекул и изменений их в процессе химических реакций.
“Физическая химия — наука, которая должна на основании положений и опытов физических объяснить причину того, что происходит через химические операции в сложных телах”. Такое определение даёт М. В. Ломоносов в своём “Курсе истинной физической химии” (1752 г.). Как и во многих других случаях, он опередил современную ему науку более чем на столетие. Следующий по времени курс физической химии читался Н. Н. Бекетовым (1865 г.). Важность данной дисциплины была широко осознана лишь к концу XIХ в.
Вопрос о внутреннем строении атомов и молекул интересовал уже М. В. Ломоносова. “Bо тьме должны обращаться физики, а особливо химики, не зная внутреннего нечувствительных частиц строения”, — писал он, ставя перед наукой будущего те задачи, которые разрешаются в настоящее время химической физикой.
Различие между физической химией и химической физикой до известной степени условно (и вторую часто включают в первую). Вместе с тем, каждая из них может быть довольно чётко отграничена от другой: предметом физической химии (классической) является суммарное рассмотрение химических процессов, протекающих с одновременным участием множества частиц, тогда как предметом химической физики — рассмотрение отдельных частиц и взаимодействий между ними, т. е. элементарных процессов.
Атомы “не неделимы по своей природе, а неделимы только доступными нам средствами и сохраняются лишь в тех химических процессах, которые известны теперь, но могут быть разделены в новых процессах, которые будут открыты впоследствии”. Это гениальное предвидение А. М. Бутлерова (1886 г.) не было понято и принято его современниками. В сознании учёных, твёрдо стоявших на точке зрения атомистической теории, укрепилось представление об атомах, как о последних, ни при каких условиях неделимых частицах вещества. Из-за этого на несколько лет задержалось правильное истолкование важного открытия, сделанного Беккерелем в 1896 г.
Известно было, что существуют вещества, которые после предварительного освещения светятся затем некоторое время сами. Явление это называется фосфоресценцией. Изучать его можно, в частности, по действию испытуемых материалов на фотографическую пластинку. Исследуя таким образом различные вещества, Беккерель заметил, что образцы, содержащие в своём составе уран, действуют на фотографическую пластинку и без предварительного освещения.
Заинтересовавшись этими опытами и продолжая их, М. Сколдовская-Кюри обратила внимание на то, что действие на фотографическую пластинку природных руд урана сильнее, чем чистой его окиси, несмотря на большее процентное содержание урана в последней. Это навело её на мысль, что урановые минералы содержат в своём составе какой-то неизвестный элемент, более активный, чем сам уран. В результате тщательной и кропотливой работы Кюри в 1898 г. удалось выделить из урановой руды два новых элемента — полоний и радий. Оказалось, что оба они действуют на фотографическую пластинку несравненно сильнее урана.
Само явление, изучавшееся в дальнейшем преимущественно на соединениях радия, было названорадиоактивностью. Опыт показывал, что активность препарата определяется исключительно содержанием в нём радия и совершенно не зависит от того, в виде какого соединения он находится. Активность препарата практически не зависит также и от внешних условий: нагревание или охлаждение, действие света, электричества и т. д. не оказывают на неё сколь-нибудь заметного влияния. Все эти факторы заставляли сделать предположение, в корне противоречащее установившимся взглядам, — предположение, что радиоактивные явления обязаны своим происхождением самопроизвольному распаду атомов радия и других радиоактивных элементов. Тем самым был поставлен вопрос о внутреннем строении атома.
Исследование радиоактивного излучения показало, что оно является сложным. Если радиоактивный препарат, заключённый в непроницаемую для его лучей свинцовую капсулу с отверстием наверху, поместить в электрическое поле, то излучение распадается на три составные части, так называемые a, b и g–лучи. Первые отклоняются к отрицательному полюсу; они представляют собой поток частиц сравнительно большой массы, заряженных положительно. Вторые сильнее отклоняются к положительному полюсу; они слагаются из частиц очень малой массы, заряженных отрицательно. Наконец, g-лучи представляют собой волны, подобные световым лучам, но более короткие. Аналогичное расщепляющее действие на радиоактивное излучение оказывает магнитное поле. Все три вида лучей действуют на фотографическую пластинку, вызывают свечение некоторых веществ и т.д.
Ещё до открытия радиоактивности было известно, что при прокаливании металлов, а также при освещении их ультрафиолетовыми лучами поверхность металла испускает отрицательное электричество. Вопрос о природе этого электричества был выяснен опытами с катодными лучами, которые получаются при электрическом разряде в разряжённом пространстве. Для их изучения использовалась установка: стеклянный сосуд, из которого выкачан воздух, впаяны анод и катод. При разрядке между ними от катода распространяются катодные лучи, которые частично проходят сквозь узкое отверстие в аноде, затем между двумя металлическими пластинками и наконец попадают в пространство, где могут быть обнаружена при помощи фотографирования или иными путями. Если между пластинками создать электрическое поле, то лучи отклоняются в сторону пластинки, заряженной положительно,— это показывает, что сами лучи заряжены отрицательно. Изменяя условия опыта (силу поля и др.), можно изучить различие между этими лучами.
В результате подобных опытов выяснилось, что катодные лучи являются потоком отрицательно заряженных частиц с очень малой массой. Этот вывод был подтверждён дальнейшими исследованиями, причём оказалось, что частички, испускаемые металлами при их нагревании или освещении, равно как частички катодных лучей и b-лучи, представляют собой одно и то же. Частички эти были названы электронами.
До работ с катодными лучами считалось, что количество электричества может изменяться непрерывно. После этих работ стали склоняться к противоположному мнению. Уже в конце ХIХ века удалось получить приблизительно правильную оценку величины наименьшего возможного количества электричества. Этот мельчайший заряд — “атом электричества” — соответствует по величине заряду электрона. Представление об атомистической природе электричества, согласно которому каждый электрический заряд составляет целое кратное от заряда электрона (е–) с тем или иным знаком, является в настоящее время общепринятым.
Первое определение заряда электрона было произведено в 1911 г., причём метод исследования основывался на наблюдении за поведением мельчайших капелек распылённого масла в электрическом поле. Если в пространство между двумя электродами ввести небольшое число таких капелек, то за каждой из них можно следить через снабжённый шкалой микроскоп.
Под действием силы тяжести капельки опускаются вниз тем быстрее, чем они тяжелее. Следовательно, по скорости падения можно вычислить вес любой отдельной капельки.
Если теперь направить в пространство между электродами пучок электронов, часть их задержится на капельках и тем самым сообщит последним отрицательный электрический заряд. При отсутствии поля это существенно не изменит поведения капелек, и они будут продолжать медленно падать. Напротив, сообщая верхней металлической пластине достаточный положительный, а нижней отрицательный заряд, можно не только приостановить падение, но и заставить заряженные капельки подниматься вверх.
Допустим, что при некоторой напряжённости поля между пластинами та или иная капелька не движется ни вверх, ни вниз. Это значит, что электрические силы в точности уравновешивают её вес. Зная напряжённость поля и вес капельки, можно рассчитать величину имеющегося на ней заряда.
Результаты многочисленных опытов при различных размерах капелек и напряжённостях поля неизменно показывали, что заряд всегда составляет целое кратное некоторого наименьшего или просто равен ему. Такое скачкообразное изменение заряда само по себе представляет наиболее убедительное доказательство атомистической природы электричества. Очевидно, что поглощение капелькой только одного электрона и должно обусловить наименьшую величину заряда, а поглощение двух, трёх и т. д. — соответствовать целым кратным от него. Наименьшая величина заряда и отвечает, следовательно, заряду электрона (1,591.10–19 Кл). Насколько эта величина мала, видно из того, что для создания силы тока в 1 А по проводу должно ежесекундно протекать 6,25.1018 электронов.
Летящий электрон отклоняется от прямолинейного пути и электрическим, и магнитным полями. Изучение характера этих отклонений позволило установить величину отношения заряда электрона к его массе (е/m). Зная заряд, можно было затем найти и массу электрона: она равна 9,11.10–31 кг. Радиус электрона оценивается в 0,2 пм.
Опыты с нагреванием и освещением металлов показывают, что наиболее легко удаляемыми частями атомной структуры являются именно электроны. Последние заряжены отрицательно, а атом в целом нейтрален; следовательно, внутри самого атома отрицательный заряд должен как-то компенсироваться положительным.
Учитывающая это модель была предложена Томсоном (1904 г.) на основе представления о положительном заряде, равномерно распределённом во всём объёме атома и нейтрализуемом электронами, вкрапленными в это “море положительного электричества”. Она не успела подвергнуться детальной разработке, так как была опровергнута работами Резерфорда.
Резерфорд проводил опыты с a-частицами. Масса каждой из них равна 4 единицам атомного веса (тогда как масса электрона составляет лишь 1/1820 такой единицы). Заряд их положителен и по абсолютной величине равен удвоенному заряду электрона. При радиоактивном распаде атома a-частицы вылетают с большой начальной скоростью.
Узкий пучок a-частиц направлялся на тонкий металлический листочек. Следить за их дальнейшим поведением можно было, передвигая по дуге приспособление, регистрирующее a-частицы. Оказалось, что большинство a-частиц проходит сквозь листочек без отклонения, часть отклоняется на различные углы, а некоторая ничтожная доля, примерно 1 частица на каждые 10000, отскакивает почти в обратном направлении. Результаты одного из опытов с рассеиванием a-частиц листочком золота приводятся ниже:
Угол отклонения.. . 15° 30° 45° 60° 75° 105° 120° 135° 150°
Число a-частиц ... . 132000 7800 1435 477 211 70 52 43 33
Результаты этих опытов, особенно отскоки частиц обратно, невозможно истолковать на основе модели Томсона. В самом деле, летящая с большой скоростью и обладающая относительно большой массой при двойном положительном заряде a-частица может быть резко отброшена назад только в том случае, если она встретит на своём пути препятствие, обладающее большим, сконцентрированным в одном месте положительным зарядом. Распределённый по всему объёму положительный заряд таких отклонений дать не может.
Креме того, каждая a-частица на своём пути через металлический листок должна пройти сквозь множество атомов, а резкие отскоки наблюдаются лишь весьма редко. Это также заставляет предполагать, что пространство в атоме вовсе не сплошь заполнено положительным электричеством. На основании результатов опытов Резерфорда объём положительно заряженной части атома, его “ядра” оценивался примерно следующим образом. Если представить себе атом увеличенным до размеров шара с диаметром 10 м, то ядро имело бы размеры булавочной головки. Поэтому громадное большинство a-частиц и не отклоняется от прямолинейного пути, несмотря на то что каждая из них пролетает сквозь много тысяч атомов.
Диаметр атома металла составляет около 300 пм. При толщине металлического листочка в 0,1 мм (10-4 м) укладывается более 300 тыс. атомов.
Отклонения испытывают лишь a-частицы, пролетающие достаточно близко к ядру одного из встречаемых на пути атомов. При этом отскакивают обратно только те, которые прямо налетают на ядро. Подсчёт относительного числа таких отскоков и позволил оценить размеры ядра.
Опыты с a-частицами дали, однако, ещё больше — они позволили приблизительно оценить также и величину положительного заряда ядер различных атомов. В самом деле, отклонения a-частиц должны быть выражены тем сильнее, чем больше положительный заряд ярда. Результаты подсчётов показали, что этот заряд равняется наименьшему электрическому заряду (е), помноженному на число, соответствующее приблизительно половине атомного веса рассматриваемого элемента.
Основываясь на своих исследованиях, Резерфорд в 1911 г. предложил новую, “планетарную” модель, уподоблявшую атом солнечной системе. В центре должно находиться очень маленькое положительно заряженное ядро, заключающее в себе почти всю массу атома, а вокруг ядра — располагаться электроны, число которых определяется значением положительного ядра. Однако подобная система может быть устойчивой только в том случае, если электроны движутся, так как иначе они упали бы на ядро. Следовательно, электроны атома должны находиться приблизительно в таком же движении вокруг ядра, как планеты вокруг Солнца.
Правильность планетарной модели атома была вскоре подтверждена дальнейшими опытами с a — и b-частицами, пути которых стало возможным видеть и фотографировать благодаря разработанной в 1911 г. Вильсоном конденсационной камере. Принцип её действия основан на том, что при охлаждении насыщенного паром воздуха капельки тумана образуются почти исключительно вокруг посторонних частичек, особенно электрически заряженных. Конденсационная камера имеет сверху и частично с боков стеклянные стенки, а внизу поршень, при быстром выдвижении которого содержащийся в ней влажный воздух несколько охлаждается за счёт расширения. Если воздух был перед опытом тщательно освобождён от пыли, то образование тумана не наблюдается. Иначе обстоит дело при прохождении через камеру a — или b-частиц. И те и другие выбивают электроны из встречных молекул, создавая тем самым множество заряженных частиц. Вокруг последних тотчас образуются капельки тумана, ясно обозначающих весь пройденный a — или b-частицей путь.
Тяжёлая a-частица, выбивая из молекулы электрон, не изменяет своего прямолинейного движения; заметное отклонение её происходит лишь тогда, когда она пролетает вблизи ядра одного из атомов. Наоборот, лёгкая b-частица при выбивании электронов и сама изменяет свой путь (особенно, когда скорость её уменьшается). Обычным является прямолинейный путь, который заканчивается, когда скорость a-частицы уменьшается настолько, что она перестаёт выбивать электроны из встречных молекул.
Подсчёты показали, что b-частица пролетает в среднем сквозь 10 тыс. атомов, прежде чем выбивает электрон, а a-частица проходит сквозь 500 тыс. атомов, не подходя более двух или трёх раз к какому-нибудь ядру настолько близко, чтобы претерпеть заметное отклонение. Это убедительно доказывает, что ядра и электроны заполняют ничтожно малую часть занимаемого атомом пространства: фактический общий объём ядер всех атомов человеческого тела составляет лишь миллионную долю кубического миллиметра.
Атомные модели.
Планетарная модель атома имела большое принципиальное значение как новый и значительный шаг на пути познания внутренней структуры атома. Однако на первых порах она не могла выть уточнена, так как не было известно ни число, ни расположение электронов в атомах отдельных элементов.
Решение первого вопроса дали работы с так называемыми рентгеновскими лучами. В 1895 году Вильгельм Рентген изучая свойства катодных лучей, обнаружил, что те места стекляной трубки, на которые попадает поток электронов, испускает какое-то новое, действующее на фотографическую пластинку излучение, легко проходящее сквозь стекло, дерево и т. д., но сильно задерживаемое большинством металлов.
Исследование рентгеновских лучей показало, что они являются аналогичными видимому свету электромагнитными колебаниями, но характеризуются гораздо меньшими длинами волн (приблизительно 0,5-200 пм). В электромагнитном спектре рентгеновские лучи располагаются между ультрафиолетовыми и g-лучами радия, частично налагаясь на последние.
Соответствующее отдельным областям электромагнитного спектра излучение различно поглащается земной атмосферой. Весьма важно существование “окна” для сантиметровых и метровых радиоволн. Оно прежде всего позводяет принимать отражение посылаемых с Земли радиоволн от различных небесных тел. Таким путём может быть, например, с недоступной ранее точностью определено расстояние до Луны (в среднем 384 тыс. км). Вместе с тем перед радиоастрономией открывается возможность регистрации собственного радиоизлучения, идущего из различных частей Вселенной.
Благодаря большой проникающей способности рентгеновские лучи широко применяются в медицине, так как позволяют путём просвечивания и фотографирования обнаружить внутри живого организма различные дефекты (переломы костей, опухоли и т. п. Очень “жёсткие” (т. е. характеризующиеся очень малой длиной волны) рентгеновские лучи применяются также для контрольного просвечивания металлического литья с целью обнаружения в нём внутренних пустот (“раковин”).
Рентгеновские лучи возникают при ударе быстро летящих электронов об атомы элементов, входящих в состав стекла. Если применять грубое сравнение, то это можно сопоставить с падением камня в спокойную жидкость — при таком ударе на её поверхности возникают волны. Характер последних будет при данной массе камня, его скорости, размерах и т. д. зависеть также и от свойств самой жидкости и изменится с заменой, например, воды на масло. Аналогично этому при данной скорости электрона характер рентгеновских лучей — их длина волны — будет меняться в зависимости от того, в атом какого элемента ударяется летящий электрон.
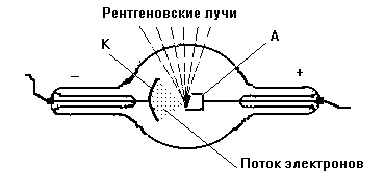
Рис. 1. Рентгеновская трубка.
Так как в состав стекла входят различные элементы, получаемое излучение содержит лучи различных длмн волн, что создаёт неудобства при пользовании им. Для избежания этого в рентгеновской трубке (рис. 1) против катода (К) устанавливается анод (А), сделанный из какого-дибо простого вещества. Попадая на его однородную поверхность, поток электронов вызывает образование рентгеновских лучей, характеризующихся некоторой определённой длиной волны.
В 1912 г. Мозли поставил перед собой задачу изучить длины волн рентгеновских лучей, получаемых от анодов, сделанных из различных химических элементов. Оказалось, что длины волн изменяются довольно закономерно, как это видно из рис. 2. При обработке результатов измерений обнаружилось, что корень квадратный из обратных значений длин волн является линейной функцией атомного номера, т. е. порядкового номера элемента в периодической системе (рис 3).
Рис. 2. Зависимость длин волн рентгеновского Рис. 3. Зависимость квадратного
излучения различных элементов от их корня из обратного значения
порядкового номера. длины волны излучения от порядкового номера элемента.
Наиболее надёжные результаты получаются при использовании жёстких рентгеновских лучей. Приметительно к ним уравнение Мозли имеет вид:
(1/l)1/2= a(Z - 1)
где l - длина волны, Z - порядковый номер элемента в периодической системе и a - константа. Неизменность этой константы при переходе от одних элементов к другим и доказывает правильность найденного соотношения.
Теоретически следовало ожидать, что длина волны должна быть тем меньше (т. е. обратное её значение тем больше), чем больше заряд атомного ядра соответствующего элемента. Результаты опытов Резерфорда показали, что заряд ядра (Z в е-единицах) равняется приблизительно половине атомного веса. Но порядковый номер, по крайней мере для не очень тяжёлых атомов, приблизительно и равняется половине атомного веса. Всё это, вместе взятое, с очевидностью указывало на то, что положительный заряд ядра численно равен порядковому номеру элемента в периодической системе.
Таким образом, каждое ядро имеет следующие основные характеристики: заряд (Z) и массу (А). В настоящее время общепринято, что структурными составляющими всех атомных ядер (“нуклонами”) являются две более простые частицы с почти одинаковой атомной массой, близкой к 1 а.е.м. Одна из них протон (р) — несёт единицу положительного заряда, а другая — нейтрон (n) — электрически нейтральна. Структуру любого атомного ядра можно выразить простой формулой Zp + (A - Z)n, где А — округлённая до ближайшего целого числа масса атома в единицах атомных масс. Например, ядро атома фтора (Z = 9, A = 19) состоит из 9 протонов и 10 нейтронов.
У большинства химических элементов ядра отдельных атомов при постоянном числе протонов (Z) могут несколько различаться числом нейтронов (A - Z). Например, ядра атомов углерода всегда содержат 6 протонов, но нейтронов могут содержать либо 6, либо 7. Поэтому в природе существуют атомы углерода с массовым числом 12 (сокращённо 12С), и с массовым числом 13 (13С). Такие атомы одного и того же элемента, характеризующиеся различными массовыми числами (т. е. суммарным числом нуклонов), носят названиеизотопов данного элемента. Обычный углерод, имеющий атомный вес 12,011, представляет собой смесь 12С (около 98,9 %) и 13С (около 1,1 %). Химические свойства изотопов практически тождественны, состав их природной смеси при реакциях обычно не изменяется.
Атом в целом электронейтрален, т. к. число электронов, входящих в структуру электронной оболочки, равно заряду ядра, т. е. порядковому (атомному) номеру соответствующего химического элемента. Установление этого числа (Z) позволило перейти к построению атомных моделей.
В общих чертах вопрос был решён Бором (1913 г.). Для химии наиболее интересны модели, разработанные в 1916 г. Косселем. Хотя при их построении принимался во внимание ряд различных свойств атомов, здесь можно ограничиться рассмотрением химической стороны рассуждений.
При переходе от лёгких ко всё более тяжёлым атомам заряды их ядер последовательно возрастают. С другой стороны, химические свойства элементов при том же переходе меняются периодически. Отсюда следует, что химические свойства определяются не столько общим числом электронов в атоме, сколько их относительным расположением.
Но если это так, то и обратно исходя из химических свойств можно получить указание на расположение электронов. В частности, следует ожидать некоторую периодичность его изменения при последовательном возрастании зарядов ядер.
Интересно было, что при определённых условиях молекула, например, поваренной соли способна распадаться на натрий и хлор таким образом, что первый оказывается заряженным положительно, а второй отрицательно. Исследование этих частиц показывает, что заряд каждой из них численно равен заряду электрона. Происхождение обоих зарядов естественнее всего объяснить переходом одного электрона с атоманатрия на атом хлора. Но в поваренной соли и натрий и хлор одновалентны — из этого следует, что одна единица валентности отвечает одному переданному электрону. Тогда в случае, например, двухвалентного кальция можно ожидать перехода двух электронов. Действительно, опыт показавает, что получающаяся в тех же условиях частица кальция имеет два положительных заряда. Точно так же и в других случаях валентность элементов совпадает с числом передаваемых электронов. Такими легче всего передаваемыми — валентными — могут быть только электроны, наиболее удалённые от положительно заряженного атома.
Наконец, большую роль играли соображения, связанные со свойствами инертных газов: то лбстоятельство, что элементы этой группы не вступали в химические реакции, указывало на особую устойчивость электронных структур их атомов.
Построение простейшей модели атома водорода не представляет трудностей: электроно вращается в этом атоме вокруг протона. Для следующего элемента — гелия — возможны уже две различные модели (рис.4): два его электрона могут вращаться по орбитам, расположенным либо на различных расстояниях от ядра (А), либо на одинаковом, что схематически обозначенопомещением их на одну окружность. Выбор между ними может быть произведён на основании химических свойств гелия. Если бы верна была модель А, то внешний электрон был бы связан в гелии не прочнее, чем в водороде. В соответствии с этим гелий должен был бы походить по свойствам на водород. Между тем он химически инертен. Это говорит за то, что оба его электрона находятся в одинаковых условиях и оба весьма прочно связаны с ядром, что и заставляет остановиться на модели Б.
Следующий элемент — литий —имеет уже три иэлектрона. Для него мыслимы четыре различные модели, коказанные на рис 5. Литий представляет собой металл, по химическим свойствам похожий на натрий и во всех своих соединениях одновалентный. Очевидно, что этому лучше всего соответствует модель Г. Принципиально важно то обстоятельство, что в ней сохраняется устойчивая конфигурация гелия из двух электронов в первом слое около ядра.
Элемент с атомным номером 4 — бериллий — всегда двухвалентен. Это показывает, что валентными являются в нём только два электрона, причём оба они находятся в одинаковых условиях. Очевидно, что и в бериллии сохраняется устойчивая гелийная двойка, а два остальных электрона располагаются в следующем слое.
Элемент № 5 — бор — трёхвалентен. Его модель, следовательно, строится аналогично модели бериллия, с той лишь разницей, что во втором от ядра слое содержится уже три электрона. Элемент № 6 — углерод — четырёхвалентен и расположение его электронов будет: 2 в первом слое и 4 во втором. Общая тенденция м развития атомных структур уже видна: при сохранении гелийной двойки в первом слое постепенно заполняется электронами второй. Это заполнение второго слоя будет, очевидно, продолжаться до тех пор, пока не достигнется число электронов, соответствующее его максимальной устойчивости. Но тогда должен получиться атом инертного газа. Рассматривая элементы, следующие в системе за углеродом, находим, что азот (2 и 5), кислород (2 и 6) и фтор (2 и 7) являются химически активными. Лишь элемент № 10 — неон — со структурой 2 и 8 оказывается аналогом гелия — инертным газом. Отсюда можно сделать вывод, что второй электронный слой становится устойчивым при 8 электронах.
Продолжая рассмотрение, находим, что элемент № 11 — натрий — одновалентен, магний — двухвалентен и т. д. Так как второй электронный слой заполнен уже в неоне, валентные электроны этих элементов будут располагаться в третьем слое.
Ввиду того, что пользование моделями атомов для выражения структур химических соединений затруднительно (с чисто графической стороны), обычно применяется упрощённый способ их изображения, при котором указывается только число электронов во внешнем слое:
Теория водородного атома
Вопрос о структуре простейшего атома — атома водорода — был разрешён в 1911 г. планетарной моделью, однако в самой этой модели таились внутренние противоречия. Действительно, по представлениям классической электродинамики вращающийся вокруг ядра электрон должен был непрерывно излучать энергию в виде электромагнитного излучения. Отсюда вытекали два важных следствия:
1. Из-за постоянного излучения энергии радиус орбиты электрона должен последовательно уменьшаться, в конце концов электрон должен упасть на ядро, что привело бы к уничтожению атомая. как такового.
2. Вследствие постепенного изменения скорости вращения электрона электромагнитное излучение атома должно состоять из непрерывного ряда лучей различных длин волн. Иначе говоря, спектр водорода может быть сплошным, т. е. содержать линии, соответствующие всевозможным длинам волн.
Ни то, ни другое следствие не оправдывается: самоуничтожения атомов водорода не происходит, а видимый спектр этого элемента состоит из ряда отдельных линий, соответствующих некоторым определенным длинам волн, как это видно из рис. III-21.
Голубой Фиолетовый
500 550 450 400 350 нм
Рис. Ill-21. Видимый спектр водорода (серия Бальмера).
Таким образом, либо планетарная модель, либо классическая теория должна была быть неправильна. На самом деле в серьезных поправках нуждались и та, и другая.
Еще до появления планетарной модели атома был отвергнут тезис классической электромагнитной теории света о непрерывности излучения. «Тезису, гласящему, что скачков не бывает, а есть только н е п р е р ы в н о с т ь, с полным правом можно противопоставить а н т и- т е з и с, по смыслу которого в действительности и з м е н е н и е в с е г д а с о в е р ш а е т с я с к а ч к а м и, н о т о л ь к о р я д м е л к и х и б ы с т р о следующих один за другим скачков сливается для нас в один «непрерывный процесс» (Плеханов). Таким антитезисом явилась квантовая теория (Планк, 1900 г.).
Согласно этой теории, энергия излучается не непрерывно, а определенными порциями, являющимися кратными некоторого «кванта действия» (h). Величина излучаемого кванта энергии тем больше, чем больше частота колебаний излучения, т. е. чем меньше длина его волны. Например, фиолетовые лучи имеют бульшую энергию, чем красные. В электромагнитном спектре (рис. III-12) наибольшей энергией обладают g-лучи, наименьшей — радиоволны. Величину кванта энергии (E в Дж) для любого электромагнитного излучения можно вычислить из соотношения Е = hn, где h — квант действия (6,62·1034Дж·с)и n — частота колебаний рассматриваемого излучения. Квантовая теория подтверждена обширным опытным материалом и является в настоящее время общепринятой.
1-2
Исходя из планетарных представлений и квантовой теории, Бор в 1913 г. построил модель атома водорода, не заключающую в себе тех противоречий, о которых говорилось выше. Модель эта была разработана на основе следующих положений.
1. Электрон может вращаться вокруг ядра не по всевозможным орбитам, а лишь по некоторым определенным. На таких «дозволенных» орбитах он вращается, н е и з л у ч а я э н е р г и и.
2. Ближайшая к ядру орбита соответствует наиболее устойчивому («н о р м а л ьл о м у») состоянию атома. При сообщении последнему энергии извне электрон может перейти на одну из более удаленных орбит, причем запас его энергии будет тем больше, чем дальше от ядра орбита, на которую он переходит, такой электрон находится на более высоком энергетическом уровне. Атом, содержащий электрон на одном из высоких энергетических уровней, в отличие от нормального, называют «возбужденным». Как показывает опыт, обратный переход из возбужденного состояния в нормальное осуществляется весьма быстро: средняя «продолжительность жизни» большинства возбужденных атомов оценивается величинами порядка 10-8 сек.
3. Поглощение и излучение атомом энергии имеет место только при п е р е с к о к е электрона с одной орбиты на другую. При этом разность энергий начального (Ео) и конечного (Ек) состояний воспринимается или отдается в виде кванта лучистой энергии (фотона), отвечающего излучению с частотой колебаний, определяемой рис. 111-22. Возможные соотношением hn = Eо- Eк.
Изложенные представления позволили вычислить радиусы различных «дозволенных» квантовыми условиями орбит электрона в атоме водорода. Оказалось, что они относятся друг к другу как 12:22:32:42:...:n2. Величина nбыла названа г л а в н ы м к в а н т о в ы м ч и с л о м. Как видно из приведенного выше, nможет принимать различные значения, соответствующие натуральному ряду целых чисел.
Радиус ближайшей к ядру орбиты (n = 1) оказался для водорода равным 53 пм. Электрон вращается по ней со скоростью около 2200 км/с(средняя скорость вращения Земли вокруг Солнца составляет 30 км/с). На рис. III-22 дана схема возможных для атома водорода орбит, причем приведены лишь первые четыре. Скорость вращения электрона на второй из них вдвое меньше, чем на второй из них вдвое меньше, чем на первой, на третьей — втрое меньше и т. д. 
Рис. III-22. Возможные электронные орбиты атома водорода по Бору
Рис. III-23. Схема происхождения водородного спектра
Работа, которую необходимо затратить для вырывания электрона водородного атома с той или иной орбиты, обратно пропорциональна квадрату ее главного квантового числа. Поэтому, например, вырвать электрон с третьей орбиты в девять раз легче, чем с первой.
Вычисленные частоты излучений, возникающих при перескоках электрона с одних орбит на другие, оказались совпадающими с частотами линий наблюдаемого на опыте водородного спектра. Как видно из рис. III-23, перескокам с различных более удаленных от ядра орбит на отвечающую n= 1 соответствуют линии серии, лежащей в ультрафиолетовой области, перескокам на орбиту с n = 2— линии серии Бальмера (рис. III-21), а перескокам на орбиты с n = 3, 4 и 5 — линии трех серий, лежащих в инфракрасной области. Две последние серии были обнаружены экспериментально уже после разработки теории водородного атома и именно на основе ее предсказаний.
5-6
Если сообщить водородному атому достаточную энергию, то происходит его ионизация — распад на электрон и протон. Энергия, которую нужно для этого затратить, отвечает n= Ґ (рис. III -24) и называется энергией ионизации (I). Она определена из спектра и для нормального состояния атома водорода составляет 1311 кДж на моль.
Н + 1311 кДж = Н+ + е
По соотношению I= 1311/n2 энергия ионизации может быть рассчитана и для возбужденных состояний атома водорода.
7
Дальнейшее развитие теории водородного атома было дано Зоммерфельдом (1916 г.), показавшим, что кроме круговых орбит электрон может двигаться и по эллиптическим (с ядром в одном из фокусов эллипса), причем почти одинаковому уровню энергии соответствует столько возможных типов орбит, сколько единиц в главном квантовом числе. Последнее определяет размер большой полуоси данного семейства эллипсов (в частном случае круга его радиус). Величина малой полуоси определяется «п о б о ч н ы м» квантовым числом (6), которое также принимает значения последовательных целых чисел, но не может быть больше главного.
Для большой полуоси эллипса действительно соотношение а = n2r, а для малой b = nkr, где r — радиус орбиты при нормальном состоянии атома (53 пм). Например, для главного квантового числа 3 возможны три типа эллипсов, характеризующиеся обозначениями 31, 32 и 33, которые показывают, что большая полуось относится к малой соответственно как 3: 1, 3: 2 и 3: 3. В последнем случае имеем частный вид эллипса — круг, который один только и рассматривался первоначальной теорией.
Модель возможных электронных орбит атома водорода по Зом- мерфельду показана на рис. III-25. Отвечающие каждой из них энергетические уровни (п о д у р о в н и) схематически сопоставлены на рис. III-26 (Б) с уровнями, соответствующими только круговым орбитам (Ф). Произведенное Зоммерфельдом уточнение модели водородного атома позволило объяснить тонкую структуру спектральных линий.
На рис. 111-26 видно, что наинизшие подуровни отвечают наиболее
**»У™ *** «Р*»™ Ри**Ш-25. Именно они и будут поэтому в первую тронных орбит атома водорода по
Зоммерфельду.
очередь заполняться электронами при построении нового слоя в м н о г о э л е-
ктронных атомах. Сами электронные слои (т. е. совокупности электронов с одинаковым значением главного квантового числа) в порядке удаления от ядра часто обозначаются буквами *, L, М, Л*, О, Р, Q.
У тяжелых атомов линии видимого спектра обусловливаются перескоками лишь самых внешних электронов, тогда как при перескоках в более глубоких слоях получаются линии, отвечающие ультрафиолето-
§ 4. «Теория водородного атома
вым или рентгеновским лучам. Энергия ионизации для этих атомов понимается как энергия, необходимая для удаления наименее прочно связанного электрона, каковым является один из занимающих самые внешние орбиты.*
Работу отрыва электрона от атома часто выражают путем указания его ионизационного потенциала. Под последним понимается то минимальное напряжение электрического поля в вольтах, при котором ускоряемый этим полем свободный электрон становится способным вызывать ионизацию данного атома (выбивая его внешний электрон). Например, ионизационный потенциал атома водорода равен 13,595 е.
Ионизационному потенциалу численно равна энергия ионизации, измеряемая в электрон-вольтах (эе), а переход от них к тепловым единицам дается соотношением: эв = 23,06 ккал/моль. Переводной коэффициент представляет собой энергию моля (т. е. 6.02.10*) электронов, приобретаемую им при прохождении ускоряющего поля с напряжением в 1 0. Таким образом, работы ионизации атомов могут быть по желанию выражены и в тепловых единицах (ккал/моль), и в электрон-вольтах (Э0).*
Рассмотренные выше представления не противоречат простейшим атомным моделям (рис. Ill-19), а лишь уточняют их. Действительно, распределение электронов по
А Б Рис. Ill-26. Схемы относительных энергетических уровней круговых и эллиптических орбит.
слоям сохраняется в моделях Бора-Зоммерфельда и соответствует приводившемуся в предыдущем параграфе. И те, и другие модели, конечно, не отображают структуру атомов во всей ее сложности. Несомненно, однако, что они все же дают правильное представление о некоторых основных чертах этой структуры. Именно так и надо их понимать. «Признание теории снимком, приблизительной копией с объективной реальности, — в этом и состоит материализм» (Л е н и н). *-*
Дополнения
1) Числовая связь между значениями длин волн, частот колебаний и энергий электромагнитного излучения для видимой части спектра (4000-7000A) и ближайших к ней областей наглядно показана на рис. 111-27. В последней включены также наиболее употребительные в химии значения соответствующих энергий в ккал на грамм- атом (т. е. на 6,02«10** фотонов). Как легко установить по рис. 111-27, энергия излучения на протяжении видимого спектра изменяется почти вдвое. '
2) При рассмотрении вопросов, связанных со спектрами, часто пользуются не непосредственно длинами волн, а их обратными значениями — т. н. волновыми чис- л а м и' а) = т,, Так как длины волн при этом выражают в сантиметрах, ш имеет рязмерность см.-*. Волновое число показывает, сколько волн данной длины укладывается на протяжении 1 см. Взаимосвязь между энергией излучения и его волновым числом хорошо передается простым соотношением: Q = ю/350 ккал/е-атом. Подобное же соотношение Q== 1/350?* (где l, выражено в см) может быть использовано для приближенного расчета энергий излучения по длинам волн. Следует отметить, что волновые числа нередко называют «частотами» и обозначают через v. Это может повести
Ill. Основные представления о внутреннем строении вещества
к недоразумениям, так как в действительности v = (ri'c, где с — скорость света. Менее опасно в этом отношении также применяемое для волновых чисел обозначеяне v. Для сбозкачения см'* иногда вводят термин «кайзер» (К), а для 1000 cjh-*-«килокай- зср» (кК).
3) Условием равновесия в круговом движении является равенство сил центробежной и центростремительной. Для атома водорода первая из них определяется энергией движения электрона и радиусом окружности, по которой он вращается, вторая- электростатическим притяжением электрона к ядру. Если /я — масса электрона
Рис. Ill-27. Длины волн и анергий излучения.
(9,1110-* г), е-его заряд (4,80-10-'° абсолютных электростатических единиц), r- радиус орбиты и и — скорость электрона, то условие равновесия для атома водорода выражается соотношением mv' e'
-»*«r*
Пмеп это одно уравнение с двумя неизвестными (и и r), еще нельзя сказать о внутренней структуре атома водорода ничего определенного.
5ор вышел из затруднения, приняв на основе представлений квантовой теории, что момент количества движения (тиг) электрона может изменяться лишь скач- к а к и в соответствии с уравнением
* * д (и== If 2) 3f...) *л,
1,054. 10--* эрг сек, -»' постоянную Планка — часто обозначают знач-
/ho2= —
2л,
Величину
ком и.
Сочетание введенного таким образом второго уравнения с предыдущим позволяет получить для обоих неизвестных параметров «движения электрона уже опреде- л е н н ы б общие решения:
h'3 _ 2rte' 4л*е*/71 " h д
Подстановка в эти выражения известных значений констант (я, и, ё, т) приводит к следующим простым расчетным формулам для радиусов «дозволенных» орбит и скоростей вращения электрона:
т (А) == 0,53д' и о == — км/сея
На орбите с д = 1 электрон совершает один оборот за время порядка К)-* сек.
4) Радиус первой электронной орбиты атома водорода входит в т. н. атомную си- cwmy единиц'. длины (0.53-10-® см), массы (9*10-* г), з9ряда (4,8.10-Ч' абс. ал. ед.), времени (2,4210-'* сек), скорости (2,2«)(? см/сек), частоты (4,1.1016 сек-*), энергии (4,36.1011 эрг, или 27,2 за, вли 2*.108 сл-'* или 627,2 кк, ал1молъ). При рассмотрении атомных объектов в такой (преДложевной Харт- р и) системе единиц уравнения часто освобождаются от числовьш множителей и приобретают более простой вид.
§ 4. Теория водородного атома
5) Потенциальная энергия двух численно равных разноименных зарядов е, находящихся на расстоянии т друг от друга, определяется выражением --. С другой
стороны, кинетическая энергия *-g-* электрона в атоме водорода равн* * (ср.
доп. 3). Так как общая энергия (Е) слагается из кинетической и потенциальной, для аточа водорода имеем * e' e' e'
Величина светового кванта (hv). отвечающего перескоку электрона в атоме водород? с одной орбиты на другую, определяется разностью энергий его начального (Ёа) и кснечного (Ек) состояний:
/м? == Ец — Ек == — -* + -* *'и. *'к.
*\**
Замена r его общим выражением (доп. 3) дает
*==**--*)
"* rl*r)
Подстановка значений констант приводит уравнение для частот колебаний к следующему расчетному виду:
v == 3,30 101* f-- — -*
*tt* п*н)
Наконец, соотношение *v = с (ср. 11 § 2) позволяет перейти от частот к длинам волн. Если выражать их в ангстремах, то расчетная форма уравнения приобретает вид:
*=*(*-*
Ниже в качестве.примера сопоставлены вычисленные по последней формуле и экспериментально определенные длины волн основных линий серии Бальмера (в ангстремах):
л™ Нд HP Hv на Теория: 6544 4848 4329 4091 Опыт; 6563 4861 4340 4102
Приведенное сопоставление показывает, что теория водор6дного атома даже в ее простейшей форме дает прекрасно согласующиеся с опытом результаты.
6) Сравнительно недавно инфракрасная часть водородного спектра была изучена более детально. Обнаружены две дополнительные линии первой серии и по одной во второй и третьей сериях. Впервые выявлена отвечающая перескоку электрона на орбиту с п = 6 четвертая инфракрасная серия, представленная линией с длиной волны 123fi84A (т. е. уже более 0,01 мм). Энергия такого излучения составляет лишь 2,3 ккал/г-атом.
7) Приводившееся выше теоретическое выражение для /м? позволяет производить различные приближенные расчеты, связанные с изменением энергетического состояния атома водорода. Вводя в уравнение множитель 1,4410*, служащий для перехода от эргов на один атом к /скал на грамм-атом, получаем
Пусть, например, требуется рассчитать энергии возбуждения, отвечай- щие линиям серии Бальмера (ив = 2). Подставляя в уравнение последовательно дв li= З* 4, 5, 6, получим:
Энергия возбуждения, ккал. ».
«а „р “v 44 59 66
70
tJ!.. Основные представления о внутреннем строении вещества
Как рядно уже из приведенного ряда цифр, по мере удаления электрона от разница между энергиями последовательного возбуждения быстро уме шается. Этим и обусловлено наблюдающееся в спектре водорода быстрое сбл] ние отдельных линий при подходе к г р а н и ц е с е р и и (ср. рис. 111-21).
Сама подобная граница соответствует як = оо, т. е. полному отрыву элект* от ядра или ионизации атома. В зависимости отян соответствующие значе энергии будут, очевидно, различными. Наиболее важна из них энергия, отвечают н о р м а л ь н о м у исходному состоянию атома («н == 1), которая обычно и ука вар-тся под названием энергии ионизации. Экспериментальное ее определен из границы ультрафиолетовой серии приводит к значению 313,6 ккал, почти не от* чающемуся от вычисляемого по приведенной выше теоретической формуле (314 кка. Величина эта, под названием ридберг (Ry), иногда принимаемся за единицу энергг Она равна половине атомной единицы (доп. 4).
8) Для отрыва последнего электрона от атомного ядра с зарядом 1 требует. затратить в У раз больше энергии, чем для ионизации атома водорода. По расчеч на грамм-атом эта энергия равна 313,6 Z* ккал. Радиусы *-слоев в сложных атома относятся друг к другу, как обратные значения зарядов ядер, т. е. с возрастание, атомного номера элемента последовательно уменьшаются. Однако даже у наиболе тяжелых атомов они все еще в сотни раз превышают собственные размеры атомны. ядер.
9) Находящийся в электрическом поле электрон отталкивается от отрицательного полюса и притягивается к положительному. Если f — разность потенциалов ускоряющего поля (в б), то создаваемая им скорость электрона определяется соотношением o=60fr*jF ка/сек. Следовательно, меняя напряжение, можно сообщать электрону определенные скорости, а тем самым и определенные величины кинетической энергии. Полезно запомнить следующее энергетическое соотношение между электрон- вольтами и волновыми числами (доп. 2): 1 эв = 8066 с.м-*.
10) Соотношение между числовыми значениями ионизационных потенциалов и энергий ионизации наглядно показано на рис. 111-28. Приводимыа в литературе значения ионизационных потенциалов, как правило, относятся к О „К. Приближенный пересчет соответствующих им энергий ионизации на 25°С может быть осуществлен путем
10 9 8 1 >t 1
5 4 3 ? fltlltlt ltfllllf 11 Ill t t t t 11 t I t I 111) t
Q I 300 250 200 W №0903070 60 'SO W 30 23кк!1л1мол{,
Рис. Ill-28. Ионизационные потенциалы и энергии ионизации.
добавления к приводимым значениям по 0,07 эв (или 1,5 ккал/моль) на каждый отрываемый электрон.
11) Ниже в качестве примера даются значения энергий ионп*а** (“i *l*i.*;* щих последовательному отрыву электронов из внешних электронных слоев атомов инертных газов. Главное квантовое число слоев указано при обозначении элемента.
Отрываемый электрон
Не (n=l) Ne (tt=2) Аг (п=5) Кг (rt=4) Хе (п=5) Rn (n=6)
24.581 21,559 15,755 13,996 12,127 10,746
54,403 41,07 27,62 24,56 21,2 *20.02)
63,5 40.90 36,9 32.1 (29,78)
97,02 59.79 (52,1) (45.46) (43,78)
126,3 75.0 (65,9) (56,9) (55,1)
157.91 91.3 (79,6) (68.3) (66,8)
(206.6) 124,0 (109,6) (96,0) (96,7)
ll*lt, (127,3) (110,4) (111.2)
Все цифры приводятся с тем числом знаков, которое отвечает предполагаемой точности их определения из спектров или расчетным путем. Такие расчеты были произведены почти для всех элементов. Результаты их, как менее надежные, здесь и далее даются в скобках.
§ 4. Теория водородного атома.
Рассмотрение приведенных данных показывает, что по мере роста главного квантового числа электронного слоя, т. е. удаления его от ядра, отрыв однотипного (например, первого) электрона последовательно облегчается. Отрыв каждого последующего электрона из одного и того же слоя требует значительно большей затраты энергии, чем отрыв предыдущего. Особенно резкий скачок наблюдается при переходе от одного электронного слоя к другому. Например, энергия ионизации аргона, соответ* ствуюшая отрыву девятого электрона (т. е. первого из слоя с n == 2), составляет 421 эв, что почти в три раза превышает значение для восьмого электрона (т. е. последнего из слоя с п = 3).
12) Начиная с середины 20-х годов текущего века в развитии учения о строении атомов наметился перелом, обусловленный влиянием новой физической концепции (т. е. познавательной идеи), выдвинутой в *924 г. де-Бройлем… Если еще из самой квантовой теории вытекало и путем изучения столкновений фотонов с электронами было экспериментально подтверждено, что к а ж д а я э л е к т р о м а г н и т н а я волна одновременно обладает свойствами частицы, то, по де-Бройлю, имеет место и обратное: каждая
движущаяся частица одновременно обладает свойствами волны.
Количественную взаимозависимость между волновыми и корпускулярными (т. е. отвечающими частицам) свойствами материи дает уравнение де-Бройля'.
l,»hinlv
17=J н»/
\*\*
*\
7.5 10 W /7=J h=Z
2.5 2.5 5 2.5 5 7.5 to и
Расстояние от ядра, *
где /I — квант действия, та — масса частицы, о — ее скорость и ?* — соответствующая длина йодны. Пользуясь этим уравнением, можно подсчитать массу кванта лучистой энергии
(u=c==3,00'10*'* см/сек), отвечающего любой длине волны. Вместе с тем можно вычислить длину волны, характерной для частицы с любой заданной массой и скоростью. Например, отвечающий линии Но, серии Бальмера (3*=6563A== 1-=6,563-10-* см) фотон имеет массу ftl=3.10--* а, т. е. он примерно в ЗООООО раз легче электрона. С другой стороны,, обладающий скоростью, например, G'K? см/сек электрон характеризуется волной с l* = 1,21 10-8 см = 1,21 А, т. е. волной типа рентгеновских лучей.
Это следствие теории вскоре нашло прямое экспериментальное подтверждение: оказзлось, что направленный на кристалл пучок электронов испытывает дифракцию подобно рентгеновским лучам. Немного позднее то же самое было установлено для атомов водорода и гелия. Так как дифракция является характерным свойством в о л н, приведенные результаты убедительно подтверждают правильность рассматриваемых представлений.
13) Развивавшаяся на базе этих представлений волновая механика подходит к вопросу о строении атомов с точки зрения характерного для нее принципа неоп- р е д е л е н н о с т и (Гейзенберг, 1925 г.). Согласно последнему характер движения электрона принципиально не может быть точно фиксирован. Модельное представление об атоме с его определенными орбитами электронов должно быть поэтому заменено описанием, при котором оценивается лишь вероятность нахождения электрона в том или ином месте пространства. Сама оценка этой вероятности производится хотя и с учетом структурных данных, но чисто математическим путем, при помощи т. н. волнового уравнения (Шредингер, 1926 г.). Последнее имеет характер постулата, истинность которого (в отличие от теоремы) устанавливается не выводом или прямым доказательством, а соответствием вытекающих из него следствий данным опыта.
Рис. 111-29 показывает распределение вероятностей нахождения электрона на том или ином расстоянии от ядра при различных квантовых состояниях атома водорода. Как видно из рисунка, при равенстве побочного и главного квантовых чисел (к == д)
Рис. Ill-29. Распределение вероятностей нахождения электрона в атоме водорода.
Ill. Основные представления о внутреннем строении вещества
псложения м а к с и м а л ь н ы х вероятностей приблизительно соответствуют радиусам круговых орбит теории Бора-Зоммерфельда. Для эллиптических орбит («
Подобный способ выражения вероятности нахождения электрона с помощью как бы «размазывания» его и оценки плотности получаемого т*им образом «электронного облака» особенно удобен, при волновомеханическом рассмотрении многоэлектронных атомов. Сплошная линия на рис. Ш-ЗО дает теоретически рассчитанное распределение элек- f тронной плотности для атома аргона. Как видно из рисунка, определенным электронным слоям (К, *-, М) теории Бора — Зоммерфельда отвечают максимумы кривой. Однако значительная плотность электронного облака (т. е. вероятность нахождения электрона) существует и м с ж д у слоями. Последние, таким образом, сколько-нибудь четко друг от друга не отграничиваются. Пунктиром показаны результаты проверки теоретического распределения путем расчета электронной плотности на основе
экспериментальных данных по рассеиванию аргоном электронов. Как видно из рисунка, обе кривые практически совпадают.
Волновомеханический подход к атомным проблемам позволил разрешить ряд вопросов, остававшихся ранее неясными, а также получить некоторые количественные результаты со значительно большей точностью, чем удавалось раньше. Цингами характерный для волновой механики о т к а з о т н а г л я д н о с т и сильно снижает познавательную ценность этого метода и таит в себе опасность скатиться к такому миропониманию, при котором «...»материя исчезает", остаются одни уравнения» (Ленин).
14) Необходимо подчеркнуть, что волновая механика отнюдь не исключает корпускулярную трактовку явлений. Более того, сами ее уравнения основаны на представлении об электроне, как о точечном заряде, а не зарядовом облаке. «К волновому и корпускулярному описанию следует относиться как к равноправным н дополняющим друг друга точкам зрения на один и тот же объективный процесс.)* (Борн)-
Рис. Ill-30. Распределение электронной
в это* аргона.
§ 5. Валентная связь. Вопрос о природе сил, кота* .lbarot образование химических соединений, возникал еще в начале XIX века. Однако тогда он не мог быть удовлетворительно разрешен.*
Благодаря развитию наших знаний о строении атомов мы теперь можем несколько ближе подойти к выяснению природы химического взаимодействия и лежащих в его основе причин. * ___* При этом нужно, конечно, иметь в виду, что «че- Электроток jjiqiunflmqs
ловеческое понятие причины и следствия всегда
несколько упрощает объективную связь явлений природы» (Л е н и н) —
Как известно, протекающий по замкнутому контуру (как ранее считалось,-от плюса к минусу) электрический ток создает магнитное поле, направленное в соответствии с «правилом буравчика» (рис. Ill-31). Аналогично (но с обратным направлением магнитного поля) ведет себя и вращающийся по орбите электрон. Вместе с тем имеет место и вращение его вокруг соб"
Рис. Ill-31. пр.***.* йу'
равчика,
Вторая группа периодической системы
Вторая группа отличается от других одинаковостью структуры внешнего электронного слоя у атомов всех входящих в неё элементов. С другой стороны, второй снаружи слой, оставаясь законченным, у отдельных представителей различен. Обстоятельство это налагает свой отпечаток на свойства соответствующих атомов и ионов и обусловливает разделение следующих за магнием элементов на две подгруппы: кальция и цинка.
Наличие во внешнем слое всех рассматриваемых атомов только двух электронов говорит за отсутствие тенденции к их дальнейшему присоединению. Напротив, сравнительно легко должна происходить отдача электрона с образованием максимально двухвалентных положительных ионов.
Подобно паре В-Si, бериллий в некоторых отношениях проявляет большое сходство со вторым элементом соседней группы — алюминием.
Бериллий и магний.
Первый из этих элементов принадлежит к числу довольно распространённых: на долю бериллия приходится около 0,001 % общего числа атомов земной коры. Содержание в ней магния составляет 1,4 %, и элемент этот является одним из наиболее распространённых. Кроме различных минералов и горных пород, соединения магния постоянно содержатся в водах океана, а также в растительных и животных организмах.
Элементарный бериллий впервые получен в 1828 г., магний — в 1808 г. Первый является “чистым” элементом (9Ве), а второй слагается из трёх изотопов — 24 (78,6), 25 (10,1) и 26 (11,3 %).
Строение внешних электронных оболочек атомов Ве (2s2) Mg (3s2) соответствует их нульвалентному состоянию. Возбуждение до обычного двухвалентного (2s12p1 и 3s13p1) требует затраты соответственно 263 и 259 кДж/моль.
Живое вещество содержит магний в количествах порядка сотых долей процента, а в состав хлорофилла входит до 2 % Mg. Общее содержание этого элемента в живом веществе оценивается величиной порядка 1011 т. Ещё несравненно больше находится его в океане: приблизительно 6·1016 т. При недостатке магния приостанавливается рост и развитие растений. Накапливается он преимущественно в семенах. Введение магниевых соединений в почву заметно повышает урожайность некоторых культурных растений (в частности сахарной свеклы), а в пищу кур — прочность яичных скорлуп. Для человека (особенно пожилого возраста) соединения магния важны главным образом тем, что предотвращают спазмы сосудов). Относительно велико их содержание в сушеных фруктах.
Помимо многочисленных силикатов, магний встречается на земной поверхности главным образом в виде углекислых минералов доломита СаСО3·МgCO3 и магнезита MgCO3. Первый иногда образует целые горные хребты, второй также встречается в очень больших количествах. Под слоями различных наносных пород совместно с залежами каменной соли иногда находятся и легкорастворимые минералы Mg, из которых наиболее важен карналит KCl·MgCl2·6H2O, служащий обычным исходным сырьём для получения металлического магния. Колоссальные запасы карналита имеются в Солекамске, где этот минерал залегает пластами мощностью до 100 м. Значительно реже встречается в природе минералы бериллия, важнейшим из которых является берилл Be3Al3(SiO3)6 или 3BeO·Al2O3·6SiO2.
В большинстве своих первичных минералов магний тесно связан с кремнеземом. Таковы, например, оливин (Mg, Fe)2SiO4 и реже встречающийся форстерит Mg2SiO4. На поверхности Земли он легко образует водные силикаты, примером которых может служить серпентин 3MgO·2SiO2·2H2O.
Богатые месторождения берилла (т. пл. 1650 °С) встречаются очень редко (но в одном из них были найдены отдельные кристаллы с массой до 16 г). Ещё реже встречаются минералы хрилоберилл Be(AlO2)2 и фенакит Be2SiO4. Бульшая часть бериллия земной коры распылена в качестве примесей к минералам ряда других элементов, особенно алюминия. Бериллий содержится также в глубинных осадках морей и золе некоторых каменных углей.
Различно окрашенные примесями прозрачные разновидности берилла известны как драгоценные камни. Сюда относятся тёмно-зелёные (от примеси соединений хрома) изумруды, голубые аквамарины и др. Хорошие изумруды очень редки и считались самыми дорогими из всех драгоценных камней. Как и рубины, их можно получить искусственно, но это гораздо труднее (требуется 1550 °С под давлением 150 тыс. атм). Крупнейший естественный изумруд весил 1795 карат. Кристаллы аквамарина достигают иногда гигантских размеров: самый крупный из них весил 100 кг.
В элементарной состоянии Be и Mg могут быть получены электролизом их расплавленных солей. При получении бериллия обычно пользуются смесью BeCl2 и NaCl, при получении магния — обезвоженным карналитом или хлористым магнием.
Металлический магний получают следующим образом: расплавленная соль помещается в железном сосуде, одновременно являющимся катодом. Анодом служит графитовый стержень, заключенный в мелкопористой керамической трубке, по которой удаляется получающийся при электролизе хлор. Во избежании окисления собирающегося в верхней части сосуда жидкого магния над ним пропускают медленный ток водорода. Получение 1 т металла требует затраты около 20 тыс. кВт·ч электроэнергии.
Наряду с обычным электролитическим большой интерес представляют электротермические методы получения магния, значение которых с каждым годом возрастает. Первый из них основан на обратимости реакции:
MgO + C + 640 кДж Ы CO + Mg,
равновесие которой при очень высоких температурах (выше 2000 °С) смещено вправо.
Практически процесс проводят, накаливая тесную смесь MgO (получаемую обжигом природного магнезита) с измельченным антрацитом в дуговой электрической печи. Выделяющиеся пары тотчас по выходе из печи разбавляют большим объёмом сильно охлаждённого водорода, благодаря чему температура их сразу снижается до 150-200 °С и равновесие не успевает сместиться влево. Осаждающийся в виде пыли металлический магний (содержащий примеси MgO и C) затем переплавляют. Полученный подобным образом металл характеризуется высокой чистотой (99,97 %).
При другом электротермическом методе получение магния в качестве восстановителя используется не углерод, а кремний (обычно применяют ферросилиций с содержанием не менее 75 % Si). Сырьём служит обожженный доломит, смесь которого с кремнием прокаливают под сильно уменьшенным давлением выше 120 °С. Реакция в этих условиях идёт по уравнению:
2(CaO·MgO) + Si + 518 кДж = Ca2SiO4 + 2 Mg,
причём единственным её летучим продуктом являются пары магния.
Эквимолярная смесь BeCl2 с NaCl плавится при 224 °С. Помимо электролиза, для получения металлического бериллия широко используется проводимая при постепенном нагревании до 1300 °С реакция по схеме:
BeF2 + Mg = MgF2 + Be + 184 кДж.
Такой путь рентабельнее электролитического.
Бериллий и магний представляют собой блестящие металлы, на воздухе покрывающиеся тонкой оксидной плёнкой, предающей им матовый вид и предохраняющей их от дальнейшего окисления. Важнейшие константы обоих элементов сопоставлены ниже:
Плотность, г/см3 | Температура плавления, °С | Температура кипения, °С | Электропроводность (Hg = 1) | |
| Be | 1,85 | 1283 | 2470 | 23 |
| Mg | 1,74 | 650 | 1103 | 22 |
Светло-серый бериллий довольно твёрд и хрупок, серебристо-белый магний значительно мягче и пластичнее. Оба элемента (особенно Mg) находят значительное практическое применение.
В парах бериллий, по-видимому, одноатомен, а пары магния содержат и молекулы Mg2 (энергия диссоциации которых оценивается в 29 кДж/моль). Аллотропические модификации обоих элементов неизвестны.
Непосредственное использование металлического магния довольно ограниченно (по объёму). Он расходуется в качестве отрицательного электрода при электрохимической защите от коррозии морских судов и трубопроводов, применяется в металлургии (как раскислитель), для конструирования некоторых гальванических элементов и т. д.
Из сплавов магния чаще всего применяется “магналий” и “электрон”. Первый представляет собой сплав Al с 5-30 % Mg. Магналий твёрже и прочнее чистого алюминия, легче последнего обрабатывается и полируется. Под техническим названием “электрон” понимаются сплавы вообще, в которых магналий является главной составной частью. Обычно подобные сплавы содержат Al (до 10,5), Zn (до 4,5) и Mn (до 1,7 %). Иногда в них вводят также Cu, Be, Ti и др. Обладая прекрасными механическими свойствами, “электрон” по плотности (около 1,8 г/см3) лишь немногим превышает чистый Mg. Как “магналий”, так и “электрон” на воздухе покрываются защитной оксидной плёнкой, предохраняющей их от дальнейшего окисления. Введение 0,05 % Mg в чугун резко повышает его ковкость и сопротивление разрыву. Интересно, что из сплава магния с небольшим количеством индия был получен монокристалл, способный под действием прилагаемой по определенному направлению внешней силы уже при сравнительно низких температурах в несколько раз увеличивать свою длину без разрыва. Ежегодная мировая выплавка магния составляет около 150 тыс. т.
Основным потребителем металлического бериллия, как такового, является в настоящее время ядерная энергетика. Так как бериллий меньше всех остальных устойчивых на воздухе металлов (например, в 17 раз меньше Al) задерживает рентгеновские лучи, он является незаменимым материалом для изготовления тех частей рентгеновских трубок, сквозь которые происходит выпускание лучей наружу). Для этой цели применяются пластинки из Ве толщиной 1-2 мм. Интересной особенностью металлического бериллия является исключительно большая скорость распространения в неё звука — 12,6 км/с. В жидком состоянии бериллий смешивается со многими металлами (Al, Zn, Cu, Fe, Ni и др.), но не смешивается с магнием. Обусловлено это, по-видимому, большим различием атомных радиусов Be (113) и Mg (160 пм).
Добавка бериллия и меди сильно повышает её твёрдость, прочность и химическую стойкость. У содержащего 3 % бериллия сплава сопротивление на разрыв в 4 раза больше, чем у чистой меди. Сплав последней с 2 % Ве вдвое твёрже нержавеющей стали и очень устойчив по отношению к механическим и химическим воздействиям. При содержании 0,5-1,3 % бериллия его сплав с медью имеет прекрасный золотистый цвет и отличается большой звучностью при ударе. Добавка 0,01-0,02 % бериллия к меди повышает её электропроводность. Подобным же образом малая добавка бериллия (0,005 %) к магниевым сплавал повышает их сопротивление окислению. Очень хорошие результаты даёт аналогичная алитированию обработка бериллием поверхности изделий из чугуна и стали. Присадка 1 % Ве к рессорной стали сильно повышает прочность и долговечность вырабатываемых из неё изделий. В частности, пружины из такой стали не теряют упругости даже при красном калении.
При нагревании на воздухе Be и Mg сгорают с образованием оксидов ЭО. Оба элемента легко соединяются с галогенами, а при нагревании также с серой и азотом. Реакции сопровождаются большим выделением тепла, причём магний реагирует, как правило, энергичнее бериллия. С водородом непосредственно соединяется лишь магний (при нагревании под давлением).
В компактном состоянии магний воспламеняется на воздухе около 650, бериллий — около 900 °С. Помимо оксидов ЭО, при сгорании обоих элементов на воздухе образуются нитриды Э3N2. На очень сильном выделении света при горении магния основано его применение для изготовления осветительных ракет и в фотографии (“магниевая вспышка”). В обоих случаях магний обычно смешивается с веществами, легко отдающими кислород. Ракетный осветительный состав может содержать, например, 45 % Mg, 48 — NaNO3 и 7 — связующего органического вещества.
Ниже сопоставлены теплоты образования некоторых соединений бериллия и магния, рассчитанные в кДж на моль металла:
| F | Cl | Br | I | O | S | N | |
| Ве | 506 | 247 | 184 | 84 | 301 | 117 | 96 |
| Mg | 560 | 322 | 259 | 180 | 301 | 176 | 80 |
| Отношение Be/Mg | 0,90 | 0,77 | 0,71 | 0,47 | 1 | 0,47 | 1,21 |
Из приведённых данных видно, что теплоты образования аналогичных производных бериллия и магния близки при сравнительно малых объёмах металлоидных атомов (F, O, N) и сильно расходятся при больших (Cl, Br, I, S), Так как сам атом бериллия гораздо меньше атома магния, это свидетельствует о значительной роли объёмных соотношений при образовании рассматриваемых соединений.
Вода на бериллий не действует. Магний почти не взаимодействует с холодной водой, но медленно выделяет из неё водород при кипячении. Разбавленные кислоты легко растворяются не только магний, но и бериллий. В своих соединениях оба элемента двухвалентны.
Оба элементы в соответствии со своими электродными потенциалами должны были бы разлагать воду. Однако при обычных температуре (а для Ве и при нагревании) такое разложение практически не происходит.
Обусловлено это малой растворимостью оксидов обоих элементов, образующих защитный слой на поверхности металлов. По характеру и толщине оксидной плёнки бериллий подобен алюминию. Оксидная плёнка на поверхности магния толще и рыхлее, а её защитное действие выражено гораздо слабее.
И бериллий и магний легко растворимы в разбавленных кислотах, не являющихся окислителями (HСl, H2SO4 и др.), но с HNO3 бериллий реагирует только при нагревании. В HF бериллий легко растворяется, тогда как магний практически нерастворим в ней (из-за образования на его поверхности защитного слоя малорастворимого MgF2).
Растворы сильных щелочей практически не действуют на магний, но растворяют бериллий (разбавленные при нагревании, а концентрированные уже на холоду) с образованием соответствующих бериллатов, например, по схеме:
2 NaOH + Be = Na2BeO2 + H2.
С водным раствором аммиака бериллий не реагирует. Магний с ним также почти не реагирует, но постепенно растворяется в растворе солей аммиака по схеме:
2 NH4• + Mg = Mg•• + H2 + 2 NH3.
Бериллий из солей аммония растворяется лишь в крепком растворе фторида по схеме:
Be + 4 NH4F = (NH4)2BeF4 + H2 + 2 NH3
или в растворе бифторида. Как видно из приведённых схем, химизм растворения обоих металлов различен (магний образует гидратированные катионы, а бериллий — комплексные анионы). Для ядерной энергетики существенно то обстоятельство, что бериллий не взаимодействует с расплавленным металлическим натрием.
Оксиды бериллия и магния представляют собой весьма тугоплавкие порошки. В кислотах они легко растворимы. Оксид бериллия растворим также в сильных щелочах. С водой он соединяются образуя гидроксиды Э(ОН)2.
Оксиды BeO (т. пл. 2580) и MgO (2850 °С)в парах сильно диссоциированы на элементы. Их энергии диссоциации равны 443 (Be) и 393 (Мg) кДж/моль. В отличие от MgO пар оксида вериллия содержит не только молекулы BeO и продукты их термической диссоциации, но также полимерные молекулы (BeO)n, где n = 2-6. Предполагается, что полимеры со значением n > 2 обладают циклическим строением. Сплав обоих оксидов имеет минимальную температуру плавления 1838 °С при 69 мол. % BeO.
Оба оксида растворимы в кислотах тем труднее, чем сильнее они были предварительно прокалены. Такое снижение реакционной способности обусловлено в данном случае только укрупнением кристаллов. При хранении на воздухе оксид магния постепенно поглощает влагу и CO2 переходя в Mg(OH)2 и MgCO3.
Сплавленный оксид бериллия хорошо проводит тепло и устойчив к температурным колебаниям (в меньшей степени то же относится к MgO). Он обладает большим электрическим сопротивлением даже при высоких температурах. Сделанные из него тигли выдерживают нагревание до 2000 °С и химически стойки по отношению почти ко всем металлам, кислотам (кроме HF) и растворам щелочей. Широкое применение находим BeO в ядерной энергетике. По-видимому, перспективно использование оксида бериллия (или керамических материалов на его основе) для изготовления некоторых деталей реактивных двигателей и газовых турбин.
Оксид магния изредка встречается в природе (минерал периклаз). Подучаемый прокаливанием природного магнезита MgO является исходным продуктом для изготовления различных огнеупорных изделий и искусственных строительных материалов (“ксилолит”и др.). Чистый оксид магния (“жженая магнезия”) находит применение в медицине как средство от изжоги и лёгкое слабительное. Кашица из замешенной на очищенном бензине оксиде магния может быть использована для снятия с бумаги жировых и маслянных пятен: ею смазывают пятно и дают бензину испаритося, после чего удаляют сорбировавший жир оксид магния.
В основе ксилолита лежит магнезиальный цемент, получаемый смешиванием предварительно прокалйнного при 800 °С оксида магния с 30 %-ным водным раствором MgCl2 (на 2 вес. ч. MgO лучше всего брать 1 вес. ч. безводного MgCl2). Вследствие образования полимерной структуры из атомов магния, связанных друг с другом посредством гидроксильных групп или атомов хлора, смесь через несколько часов даёт белую, очень прочную и легко порирующуюся массу.
При изготовлении ксилолита к исходной смеси примешивают опилки и т. п. Кроме ксилолита, используемого главным образом для покрытия полов, на основе магнезиального цемента готорят жернова, точильные камни и т. д.
Белые аморфные гидроксиды бериллия и магния очень малорастворимы в воде. растворенная часть Mg(OH)2 диссоциирована по типу основания, а Be(OH)2 имеет амфотерный характер и диссоциирует по суммарной схеме:
Be2+ + 2 OH-Ы Be(OH)2є H2BeO2Ы 2 H+ + BeO22+.
Ввиду слабости кислотных свойств Be(OH)2 соли с анионом BeO22- (бериллаты) в водном растворе очень сильно гидролизованы. Основные свойства Be(OH)2 выражены гораздо отчетливее кислотных, но всё же значительно менее, чем у Mg(OH)2, являющимся основанием средней силы. В соответствии со своим химическим характером Be(OH)2 растворяется и в сильных щелочах, и в кислотах, а гидроксид магния — только в кислотах.
Осаждение Ве(ОН)2 в процессе нейтрализации кислого раствора наступает около рН = 5,7, а Mg(OH)2 — при рН = 10,5. Наиболее надёжные значенияпроизведений растворимости этих гидроксидов в свежеосаждённом (аморфном) или окристаллизованном состояниях при 25 °С равны, по-видимому, 2·10-21 или 3·10-22 (Be) и 6·10-10 или 1·10-11 (Mg). Их вторые константы диссоциации по основному типу характеризуются значениями 3·10-8 (BeOH•) и 3·10-3 (MgOH•).
Гидрокисд магния встречается в природе (минерал брусит). Помимо кислот он растворим в растворах солей аммония (что имеет значение в аналитической химии). Растворение, например, в NH4Cl протекает по схеме:
Mg(OH)2 + 2 NH4Cl Ы MgCl2 + 2 NH4OH
и обусловлено образованием сравнительно малодиссоциированного (особенно в присутствии избытка NH4Cl) гидроксида аммония. Подобно Mg(OH)2 ведут себя и многие другие гидроксиды, хотя и мало, но всё же заметно растворимые в воде [например,Mn(OH)2]. Напротив, практически нерастворимые в воде гидроксиды, например Be(OH)2, нерастворимы и в растворах солей аммония. Однако в растворах этилендиамина гидроксид бериллия растворяется.
Для технологии бериллия важно то обстоятельство, что его гидроксид, в противоположность Al(OH)3 хорошо растворим в растворе (NH4)2CO3 (или в насыщенном растворе NaHCO3). Этим иногда пользуются для отделения Be от Al при переработке берилла. Последний первоначально сплавляют с K2CO3, и полученный сплав для осаждения кремневой кислоты обрабатывают при нагревании разбавленной H2SO4. Бульшую часть алюминия выделяют в виде кальевых квасцов, а остаток его (и примеси Fe) осаждаются раствором (NH4)2CO3. Из подкисленного HСl фильтрата кипячением удаляют CO2 и затем осаждают Be(OH)2 углекислым аммонием или перекристаллизацией и возгонкой основного ацетата бериллия.
При нагревании гидроксида бериллия на воздухе он начинает терять воду уже выше 230 °С (но полное обезвоживание достигается лишь при 500 °С. Однако при очень высоких температурах молекулы Be(OH)2 устойчивы. Поэтому прокаливание ВеО в струе водяного пара выше 1000 °С сопровождается зхаметным образованием и последующим распадом Be(OH)2, что практически сводится к возгонке оксида бериллия. В случае MgO такая летучесть с водяным паром не наблюдается.
Полная константа диссоциации H2BeO2 (на 2H• + BeO2”) оценивается значением 2·10-30. Свежеосаждённый Be(OH)2 растворяется в избытке разбавленного раствора щёлочи, но при стоянии такого раствора вновь выделяется его кристаллическая форма, которая растворима только в очень крепких (10 н.) растворах щёлочи. Выделение бериллатов возможно лишь из спиртовых растворов. Подобным путём были получены бесцветные K2BeO2 и Na2BeO2. В водной среде при отсутствии избытка щёлочи обе соли практически нацело гидролизованы. Сухим путём была получены бериллаты некоторых двухвалентных металлов.
Хотя для гидроксида магния кислотная функция совершенно нехарактерна, однако взаимодействие Mg(OH)2 с 65 %-ным раствором NaOH при 100 °С был получен бесцветный кристаллический гидроксомагнезат натрия —Na2[Mg(OH)4]. Известны и аналогичные соли типа Э2[Mg(OH)6], где Э — Sr, Ba. Все эти соединения при контакте с водой подвергаются полному гидролизу.
Для обоих элементов изместны аналогичные гидроксидам этоксидные производные Э(OC2H5)2. Лучше изученный этоксид магния может быть получен взаимодействием его амальгамы со спиртом. Он представляет собой белый порошок, растворимый в спирте и разлагаемый водой. В качестве аналога бериллатов можно рассматривать этоксидный комплекс K2[Be(OC2H5)4].
Взаимодействие свежеосаждённого Mg(OH)2 с 30 %-ной H2O2 при 0 °С был получен пероксид магния — MgO2. Он описывается как бесцветное микрокристаллическое вещество [d(OO) = 150, d(MgO) = 208 пм], малорастворимое в воде и постепенно разлагающееся при хранении на воздухе. Данные эти нельзя считать полностью достоверными: возможно, что в действительности был получен гидрат пероксида (известны 2MgO2·H2O и MgO2·H2O). Нагревание последнего из них сопровождается при 100 °С его обезвоживанием, а при 375 °С — переходом MgO2 в MgO. Теплота образования MgO2 из элементов оценивается в 623 кДж/моль. Содержащие её препараты находят применение при отбелки тканей, для дезинфекции и в медицине (при некоторых желудочно-кишечных заболеваниях). Их иногда вводят также в состав зубных порошков.
Для бериллия пероксидные соединения не получены, но сообщалось об образовании BeO2 в результате взаимодействия озона с суспензией Be(OH)2 во фреоне-12 при -65 °С. В аналогичных условиях из MgO2 частично (до 60 %) образуется надпероксид магния — Mg(O2)2. Устойчивый лишь ниже -30 °С.
Большинство солей бериллия и магния хорошо растворимо в воде. Растворы содержат бесцветные ионы Э2+. Присутствие иона Мg2+ сообщает жидкости горький, иона Be2+ — сладковатый вкус. Соли Be заметно гидролизуются водой уже при обычных температурах, соли Mg и сильных кислот — лишь при нагревании раствора. Все соединения бериллия очень ядовиты.
Возможность отравления соединениями бериллия связана главным образом с их наличием в воздухе (которое для производственных помещений не должно превышать 0,001 мг/м3). При вдыхании нерастворимых соединений главная масса этого элемента откладывается в лёгких, а при вдыхании растворимых (или их поступлении через рот) — в костях. Первыми симптомами острого отравления обычно является раздражение верхних дыхательных путей и глаз. Хроническое отравление иногда проявляется лишь через очень долгое время, причём наблюдаются общая слабость, раствойство пищеварения, одышка, кашель и серьёзные изменения в лёгких. Соединения бериллия могут вызывать также поражение костей и кожи.
Галогениды Be и Mg бесцветны. Некоторые их константы сопоставлены выше:
BeF2 | BeCl2 | BeBr2 | BeI2 | MgF2 | MgCl2 | MgBr2 | MgI2 | |
Температура плавления, °С | 821 | 425 | 509 | 480 | 1253 | 714 | 710 | 650 |
Температура кипения, °С | 1169 | 510 | 520 | 488 | 2260 | 1418 | 1430 | - |
| d(ЭГ), пм | 140 | 175 | 191 | 210 | 177 | 218 | 234 | 252 |
Приведённые значения ядерных расстояний относятся к индивидуальным молекулам рассматриваемых соединений в их парах. Молекулы эти линейны. Тенденция к их ассоциации выражена слабо: было показано, что в парах BeF2 почти не ассоциирован, а BeCl2 и галогениды магния содержат лишь около 1 % молекул (ЭГ2)2 и ещё раз в 100 меньше (ЭГ2)3. Для твёрдого BеСl2 характерно поканное на рис сочетание отдельных молекул в полимерные цепи типа ···BeCl2···BeCl2··· с мостиковыми связями бериллия через атомы хлора [d(BeCl) = 202 пм, РClBeCl = 98°, РBeClBe = 82°]. Следует отметить, что твёдрые галогениды способны существовать в различных модицикациях по некоторым характеристикам. Например, для BeF2 известна форма с т. пл. 542 °С. Расплавленные галогениды бериллия очень плохо проводят электрический ток. Для энергий решётки галогенидов MgГ2 дают следующие значения (кДж/моль): 2880 (F), 2487 (Cl), 2412 (Br), 2312 (I).
Почти все галогениды Be и Mg расплывыются на воздухе и легкорастворимы в воде. Исключением является MgF2 растворимость которого весьма мала (0,08 г/л). В растворах мало диссоциированы лишь ионы BeF• (К = 5·10-5) и MgF• (К=5·10-2). Подвижность иона Be•• почти вдвое меньше, чем иона Mg••. Большинство солей выделяется из растворов в виде кристаллогидратов BeCl2·4H2O, MgCl2·6H2O и т. д. При их нагревании происходит отщепление части галогенводородной кислоты и остаются труднорастворимые в воде основные соли.
Реакции присоединения характерны главным образом для фторидов, образующих комплексы преимущественно типов М[ЭГ3] и M2[ЭГ4], где М — однавалентный металл. Примерами могут служить K[BeF3] (т. пл. 406) и K[MgF3] (1070), K2[BeF4] (791) и K2MgF4] (846 °С с разл.). Особенно характерны для бериллия производные типа M2[BeF4]. По растворимости они походи на сульфаты. Ядерное расстояние d(BeF) в ионе [BeF4]2- 155 пм, а полная константа его диссоциации в растворе (на Be + 4 F’) оценивается значением 4·10-17. Известен также ряд солей, производящихся от комплексной кислотыH2[BeF3OH].
Для других галогенидов бериллия комплексообразование с соответствующими галогенидами щелочных металлов не характерно, но некоторые производные аниона [BeCl4]2- [имеющего структуру тетраэдра d(BeCl) = 189 пм] известны. Магнийдихлорид дпёт производные типа M[MgCl3]·6H2O, к числу которых относятся, в частности, природный карналит и аналогичная аммонийная соль. Нагреванием последной может быть получен безводный MgCl2 (без образования основной соли). Безводный BeCl2 легко растворяется в спирте и эфире (причём из последнего раствора соль выделяется с двумя молекулами кристаллизационного эфира). Хорошо растворимы в эфире (и ряде других кислородсодержащих органических жидкостей) также MgBr2 и MgI2. Последняя соль при взаимодействии с эфиром образует два слоя, выше 38 °С смешивающихся друг с другом. Гидролиз её при взаимодействии с водой протекает очень бурно. Получать BeI2 удобно действием сухого HI на карбид бериллия при 700 °С (с последующей очисткой посредством возгонки в вакууме). Кристаллогидрат BeBr2·4H2O может быть выделен из насыщенного бромистым водородом концентрированного водного раствора этой соли. Из роданидов бериллия были получены стеклообразный Be(NCS)2 и кристаллический Cs2[Be(CNS)3]·2H2O.
Практическое применение из галогенидов Bе и Mg находит главным образом MgCl2 большие количества которого получаются в виде отходов от переработки карналита на KСl. При 176 °С карналит плавится в своей кристаллизационной воде и одновременно разлагается, причём KСl почти полностью осаждается, а MgCl2·6H2O (т. пл. 106 °С) остаётся в виде жидкости. Хлористый магний применяется для получения из него металлического Mg, магнезиального цемента, в медицине и т. д. Медицинское применение находит также MgI2. Из растворов этой соли в метиловом спирте может быть выделен кристаллогидрат MgI2·6CH3OH.
Гидриды бериллия и магния представляют собой белые твёрдые вещества, нелетучие и имеющие характер полимеров (вероятно, образованных посредством водородных мостиков). Оба они нерастворимы в эфире и могут быть получены по схеме:
Э(СН3)2 + LiAlH4 = LiAlH2(CH3)2 + ЭH2.
При температурах порядка 400-500 °С под давлением водорода 100-200 атм и в присутствии следов иода MgH2 образуется непосредственно из элементов (теплота образования 75 кДж/моль). Оба гидрида устойчивы на сухом воздухе, но разлагается водой (BeH2 — бурно, MgH2 — медленно). Термический распад BeH2 (теплота образования 21 кДж/моль) наступает около 190, а MgH2 — около 300 °С. Подобно другим гидридам металлов, они являются сильными восстановителями.
Взаимодействие диметильных производных Be и Mg с эфирным раствором HN3 при очень низких температурах были получены азиды этих элементов — Be(N3)2 и Mg(N3)2. Они представляют собой белые твёрдые вещества. Цианомид магния может быть получен осторожным нагреванием в вакууме его диаммиаката [Mg(NH3)2](CN)2, осаждающегося при действии HCN и NH3 на концентрированный раствор Mg(NO3)2. Водой Mg(CN)2 гидролизуется, а при нагревании выше 200 °С переходит в цианамид магния — MgNCN.
Нитраты бериллия и магния легкорастворимы не только в воде, но и в спирте. Кристаллизуются они обычно в виде Be(NO3)2·4H2O (т. пл. 60) и Mg(NO3)2·6H2O (90 °С). При нагревании выше температур плавления оба нитрата отщепляют не только воду, но и HNO3, а затем переходят в сответствующие оксиды. Безводные Be(NO3)2 и Mg(NO3)2 могут быть получены осторожным нагреванием в вакууме их двойных соединений Be(NO3)2·2N2O4 и Mg(NO3)2·N2O4 (которые образуются при взаимодействии безводных хлоридов с диоксидом азота в присутствии этилацетата). Они представляют собой бесцветные твёрдые вещества.
Для сульфатов бериллия и магния характерны легкорастворимые кристаллогидраты BeSO4·4H2O и MgSO4·7H2O. Первый из них полностью обезвоживается при 400, второй — при 200 °С. Для солибериллия установлено строение [Be(OH2)4]SO4 с тетраэдрическим окружением атома Ве четырьмя молекулами воды, Подобная же структура [Be(OBe)4]SO4 вероятна и для основного сульфата бериллия, образующегося при растворении BeO в концентрированном растворе BeSO4. Интересно, что растворитель этой основной соли больше, чем самого мульфата бериллия. Раствор последнего способен растворять также металлический магний и его оксид по схемам:
2 BeSO4 + Mg + 2 H2O = H2 + (BeOH)2SO4 + MgSO4
и 2 BeSO4 + MgO + H2O = (BeOH)2SO4 + MgSO4.
Электролитическая диссоциация MgSO4 характеризуется значением константы 5·10-3. Безводный BeSO4 (т. пл. 540 °С) труднорастворим в холодной воде (но переводится в растворимое состояние горячей). Возможно, что это обусловлено его строением не по ионному, а по полимерному типу ···BeO2SO2BeO2SO2··· аналогичному структуре хлорида. Он начинает отщеплять SO3 около 580 °С, а термическое разложение MgSO4 (т. пл. 1130 °С при бычтром нагревании) становится заметным лишь выше 1000 °С.
В природе MgSO4 встречается в виде минералов горькой соли MgSO4·7H2O и кизерита MgSO4·H2O. Последний минерал даже при нагревании растворяется очень медленно (по-видимому, лишб после перехода в более богатый водой кристаллогидрат). Природный кизерит может служить хорошим материалом для получения MgO и SO2, так как при прокаливании с углём разлагается по схеме:
MgSO4 + C + 268 кДж = CO + SO2 + MgO.
Горькая соль применяется в текстильной промышленности, а также используется в медицине как слабительное.
С сульфатами некоторых одновалентных металлов BeSO4 и MgSO4 образуют двойные соли, для бериллия типа M2[Be(SO4)2], а для магния так называемые шениты состава M2[Mg(SO4)2]·6H2O, где M — одновалентный катион. Шенитом K2[Mg(SO4)2]·6H2O пользуются иногда в качестве калийного удобрения.
Почти нерастворимые в воде нормальные карбонаты ЭСO3 бериллия и магния могут быть получены лишь при одновременном присутствии в растворе большого избытка CO2. В противном случае осаждаются также почти нерастворимые основные соли [например, (ЭOH)2CO3]. Последние всегда и образуются при действии на соли Be и Mg растворами углекислых солей. Нагревание с крепким раствором KHCO3 они могут быть переведены в нормальные карбонаты, для которых характерны кристаллогидраты BeCO3·4H2O и MgCO3·3H2O. Основная соль приблизительного состава 3MgCO3·Mg(OH)2·3H2O под названием белой магнезии находит медицинское применение (при повышенной кислотности желудочного сока) и вводится иногда в состав зубных порошков.
Нормальные карбонаты Be и Mg сравнительно легко отщепляют углекислый газ и переходит в соответствующие оксиды. Для BeCO3 такой расход становится заметным уже немного выше 100, для MgCO3 — около 500 °С. На этом основано использование природного магнезита для получения СО2 и MgO. Выделенный таким путем углекислый газ очень чист и служит, в частности, для производства искусственных минеральных вод. Обожженный магнезит (т. е. в основном MgO) идет на изготовление огнеупорных кирпичей и т. п.
49) При пропускании СО2 в жидкость, содержащую взвешенный MgCO3, происходит растворение осадка, обусловленное реакцией по схеме:
MgCO3 + H2O + СO2 = Mg(HCO3)2
В кристаллическом состоянии бикарбонат магния был получен при 0 °С под давлением СО2 в 18 атм. Он очень неустойчив и на воздухе отщепляет СО2.
50) Карбонаты Be растворяются в избытке карбонатов щелочных металлов и — особенно легко — крепкого раствора (NН4)2СО3. Растворение обусловлено образованием двойных солей типа М2[Ве(СО3)2]. Аналогичные, но труднорастворимые соли дает и углекислый магний при обработке его крепкими растворами щелочных карбонатов (или бикарбонатов). Известны, например, K2[Mg(CO3)2]·4H2O, KH[Mg(CO3)2]·4Н2O и др. К солям этого типа относится и природный доломит — Ca[Mg(CO3)2]. Образованием подобного же, но легкорастворимого аммонийного производного обусловлено, вероятно, отсутствие осаждения при действии на соли магния крепким раствором (NH4)2CO3. Напротив, разбавленные растворы этого реактива в отсутствие других аммонийных солей частично осаждают основные карбонаты магния (большей частью лишь при нагревании или после долгого стояния).
51) Большое практическое применение для сушки многих жидкостей и газов находит безводный перхлорат магния («ангидрон»). Получают его обезвоживанием (в вакууме при 240 °С) кристаллогидрата Mg(CIO4)2·6H2О (т. пл. 146 °С), выделяющегося из водных растворов этой соли. По интенсивности осушающего действия ангидрон приближается к фосфорному ангидриду, имея перед последним то важное преимущество, что длительным нагреванием до 240 °С под вакуумом поглотившая воду соль может быть вновь обезвожена и повторно использована. Термическое разложение Mg(CIO4)2 наступает при 382 °С. Соль эта хорошорастворима не только в воде, но и в ряде органических растворителей.
52) Известен и перхлорат бериллия — Ве(С1О4)2·4Н2О. Подобно остальны тетрагидратам иона Be2+, соль эта имеет, вероятно, строение [Ве(ОН2)4](С1O4)2. При нагревании она не теряет воду вплоть до начала разложения аниона.
53) Нормальный ацетат бериллия — Be(СНзСОО)2 — не может быть получен из водных растворов. Он образуется, в частности, при нагревании BeCl2 с безводной уксусной кислотой. Соль эта не растворяется ни в органических растворителях, ни в воде (но медленно разлагается ею с образованием основных солей). При 295°С плавится с разложением.
54) Для хорошо растворимого в воде ацетата магния характерен кристаллогидрат Mg(CH3COO)2·4H20 (т. пл. 65°С). Растворы этой соли проводят электрический ток в несколько раз хуже, чем то обычно для солей типа ЭХ2, а при высоких концентрациях становятся очень вязкими. И то и другое обусловлено, по-видимому, полимеризацией молекул Mg(CH3COO)2, протекающей с возникновением донорно-акцепторных связей O-Mg от карбонильного кислорода анионов.
55) Оксалат бериллия — ВеС2O4·3Н2O — является единственным оксалатом двухвалентного металла, хорошо растворимым в воде. Электропроводность его разбавленного раствора примерно в 5 раз меньше, чем у BeSO4, и почти не изменяется при повышении концентрации. Такое поведение соли согласуется с трактовкой структуры по типу Ве1Ве(С2O4)21·6Н20. Вместе с тем для константы диссоциации молекулы ВеС2О4 было найдено значение 6·10!!!!. Известны и комплексные оксалаты, например Na2[Be(C2O4)2]·H2O.
56) В отличие от бериллия, оксалат магния — MgC2O4·2Н2О — малорастворим воде (0,1 г/л). Для константы электролитической диссоциации этой соли значение 4·10!!!!. В растворе (NH4)2C2O4 она растворяется с образованием довольно устойчивого комплекса (NI-l*IMg*O*l. Вместе с тем раствор щавелевой кислотыспособен растворять гораздо больше MgO, чем то соответствует реакции нейтрализации, что указывает на лёгкое образование хорошо растворимых основных оксалатов магния.
57) Для фосфата магния — Mg3(PO4)2 (т. пл. 1357 °С) — даётся значение ПР = 2·10!!!!, т. е. соль эта очень малорастворима в воде. Образованием кристаллического осадка MgNH4ЭO4 (где Э — Р или As) пользуются в аналитической химии для открытия иона Mg2+ с одной стороны, фосфорной и мышьяковой кислот — с другой. Реакция идет, например, по уравнению:
MgCl2 + NH4OH + Na2HPO4 = MgNH4PO4 + 2 NaCl + Н2О.
Фосфаты бериллия известны, но изучены хуже.
58) Боранаты бериллия и магния могут быть получены в эфирной среде по схеме:
ЭН2+В2Н6 = Э(ВН4)2.
Оба они представляют собой белые твёрдые вещества, растворимые в эфире и разлагаемые водой. Боранат бериллия летуч (т. возг. 91, разлагается выше 120 °С), тогда как боранат магния нелетуч (плавится в вакууме около 180 и разлагается лишь выше 260 °С). Молекула Ве(ВН4)2 полярна (m = 2,06). Данные трёх разных исследований её строения сильно расходятся, а Mg(BH4)3 имеет, вероятно, ионную структуру. Боранат бериллия мог бы, по-видимому, служить высококалорийной добавкой к реактивным топливам.
59) Алюмогидриды бериллия и магния были получены в эфирной среде по схеме:
ЭГ2 + 2 LiAlH4 = 2LiГ + 3 Э(AlH4)2
(где Г — С1, Вг). Они представляют собой белые твердые вещества, хорошо растворимые в эфире и разлагаемые водой. Строение их, вероятно,!!! по типу Н2А1ННЭННА1Н2. Более устойчивый Mg(AlH4)2 термически разлагается (в отсутствие воздуха) лишь выше 140 °С.
60) Из других солей рассматриваемых элементов особенно интересны комплексные производные бериллия, в которых основным центральным атомом является кислород с координационным числом 4. Типичным представителем соединений этого типа является основной ацетат бериллия ОВе4(СН3СОО)6, получаемый взаимодействием его основного карбоната с безводной СНзСООН.
Рис.. Схема пространственного строения основного ацетата бериллия.
Рис.. Валентвая схема основного ацетата бериллия.
Как видно из рис., центральный атом кислорода тетраэдрически окружён четырьмя атомами бериллия, а каждый из последних — четырьмя атомами кислорода. Помимо восьми обычных валентных связей в формировании молекулы участвуют 8 донорно-акцепторных связей О-Ве (рис. ). Получающаяся замкнутая структура так прочна, что кристаллизующийся в правильных октаэдрах основной ацетат бериллия не только плавится, но и кипит без разложения (т. пл. 285, т. кип. 331 °С). Это используется для очистки бериллия от примеси многих других элементов.
Основной ацетат нерастворим в воде, но растворяется в неполярных органических растворителях. Подобные ему производные бериллия известны и для некоторых других органических кислот. Вместе с тем, в отличие от Mg2+, А13+ и многих других катионов, Be2+ не имеет тенденции к комплексообразованию с “трилонами”. Обусловлено это, по-видимому, его очень малым радиусом.
61) Подобным основному ацетату неорганическим соединением бериллия является основной !!!!, который может быть получен термическим разложением Be(NO3)2 (в вакууме при 125 °С). Он представляет собой бесцветные летучие кристаллы, нерастворимые в неполярных органических растворителях. Известны и соли типа!!! (где M-Na, К, NH4), строение аниона которых также аналогично основному ацетату бериллия. Для магния был выделен основной ацетат состава 4Mg(CH3COO)2·Mg(OH)2, при нагревании выше 200 °С разлагающийся до MgO.
62) Сходным по свойствам с основным ацетатом производным бериллия является его ацетилацетонат — !!!!.. Он плавится при 108 °С и кипит без разложения при 270«С, почти нерастворим в воде, но хорошо растворяется в неполярных органических растворителях (СбНб, CSa и др.). Известен (но мало изучен) и ацетил- ацетонат магния.
63) Из циклопентадиенильных производных рассматриваемых элементов Ве(С5Н5)2 интересен своим строением: атом Be располагается между параллельными и отстояшими на 337 пм друг от друга плоскими кольцами C5H5 [с d(CC)= 143 н d(CH) = 110 пм], но на разных расстояниях от них [(ВеC) = 191 и 226 пм]. Возможно, что этой асимметрией и обусловлено наличие у Be(C5H5)2 дипольного момента !!!!.. Сама она связана, вероятно, с ионным характером данного!!! и малыми размерами Be2+, что создает возможность существования двух чётких минимумов на кривой его потенциальной энергии (при перемещении между кольцами).
Аналогичный по составу циклопентадиенил магния — Mg(C5H5)2 — может быть получен прямым действием паров циклопентадиена на нагретый металл. Он представляет собой бесцветное кристаллическое вещество (т. пл. 177, т. кип. 221 °С), очень чувствительное не только к воде и воздуху, но даже к углекислоте.
64) Для многих безводных солей бериллия и магния характерно легкое образование комплексных аммиакатов. При обычных температурах для них типичны составы [Be(NH3)4]X2 и [Mg(NH3)4]X2 (где X — одновалентный анион). Некоторые аммиакаты довольно устойчивы по отношению к нагреванию. Например, давление аммиака над [Be(NH3)4]Cl2 при 156 °С равно лишь 6 мм рт. ст., а над!!! оно достигает атмосферного лишь при 227 °С. Интересен гидразиновый комплекс !!!!, в котором бериллий имеет, по-видимому, необычное для него координаанонное число 6. Вероятно, таково же оно и в устойчивом лишь при низких температурах (давление аммиака достигает 90 мм рт. ст. уже при -50°С) аммиакате BeCl2·!!!!.. Водой все эти соединения разлагаются.
65) Из амидных производных Be и Mg были описаны Be(NH2)2,!!! (где M — Na, К). Известен также амидобромид Mg(NH2)Br.
Лучше других изученный Mg(NH2)2 медленно выделяется при хранении голубого раствора магния в жидком аммиаке. Он представляет собой белый порошок, самовоспламеняющийся на воздухе и бурно разлагаемый водой. При нагревании в отсутствие воздуха происходит его разложение по схеме:
3Mg(NH2)2 = Mg3N2 + 4NH3.
Из более сложных соединений того же типа интересен получаемый взаимодействием ВеН2 с диметиламином BeN(CH3)2, строение тримерной молекулы которого (образованной с участием донорно-акцепторных связей N-Be) показано на рис..
66) Сульфиды бериллия и магния, а также аналогичные им селениды и теллуриды могут быть получены прямым синтезом из элементов. Производные бериллия с водой не взаимодействуют, тогда как производные магния более или менее гидролизуются. Для теплот образования из элементов даются значения (ккал/моль): 56 (BeS), 83 (MgS), 65 (MgSe) и 50 (MgTe). Удобным способом получения чистого бесцветного MgS является пропускание тока азота с примесью паров CS2
над нагретым до 800 °С оксидом магния. При 1200 °С подобным же образом реагирует с CS2 и окись бериллия. Для магния получены также полисульфиды MgS4 и MgS5.
67) Бесцветные нитриды Be и Mg могут быть получены непосредственным соединением элементов с азотом. В случае Be реакция быстро идет начиная с 900 °С, а в случае Mg — c 500 °С. Теплоты образования нитридов равны соответственно 140 и 111 ккал/моль. Оба они относятся к соединениям ионного типа (с радиусом!!! около 140 пм). Нитрид бериллия Be3N2 очень твёрд, плавится лишь около 2200 °С (под давлением) и холодной водой почти не разлагается. Интересным путем его синтеза является протекающая при 700 °С реакция по уравнению:
3 Be + 2 KCN = Be3N2 + 2 C + 2 K.
Нитрид магния (Mg3N2) выше 700 °С начинает распадаться на элементы, при нагревании на воздухе сгорает до MgO, а водой тотчас разлагается на Mg(OH)2 и NH3. Известны также смешанные нитриды!!! (где Э — Ве, Mg) и нитрофториды магния (!!! MgaNF, MggNFg). Для аналогов Mg3N2 даются следующие значения теплот образования из элементов (ккал/моль): 128 (Мg3Р2), 96 (Mg3As2), 79 (Mg3Sb2) и 40 (Mg3Bi2). Фосфид состава Ве3Р2 известен и для бериллия.
68) Карбид бериллия состава Ве2С (т. пл. 2150 °С с разл.) образуется при накаливании ВеО с углем в электрической печи. Теплота его образования из элементов равна 22 ккал/моль. Судя по строению кристаллической решетки (обратный тип CaF2), карбид бериллия может рассматриваться как ионное соединение (Ве2+С4-). Водой он медленно разлагается с выделением метана. Образующийся при 450 °С по схеме
!!!!
карбид бериллия разлагается водой с выделением ацетилена. Он является, следовательно, типичным ацетилидом.
69) Из карбидов магния — MgC2 и Mg4C3 — первый удобно получать нагреванием порошкообразного Mg до 500 °С в токе этана, второй — до 600 °С в токе пентана. Оба карбида являются эндотермичными соединениями с теплотами образования из элементов соответственно -21 и -18 ккал/моль. Водой MgC2 разлагается с выделением ацетилена, а Mg4C3 —аллилена (СН3-СєСН) и аллена (H2C=C=CH2). Выше 550 °С MgC2 распадается на углерод и Mg2C3, который выше 750 °С разлагается на элементы. Последний карбид является единственным соединением этого класса, содержащим в своей структуре анионы [С=С=С]4+.
70) Из силицидов магния известны Mg2Si (т. пл. 1100 °С) и Mg!!!!Si!!!!.. В отличие от MgC2, теплота образования Mg2Si из элементов положительна (19 ккал/моль). Бериллий сплавляется с кремнием, но определённых соединений с ним не образует. Из аналогов силицида магния были получены !!!!MgaSn (т. пл. 780 °С) и Mg2Pb (теплоты образования из элементов соответственно 18 и 13 ккал/моль). Последнее соединение бурно реагирует с водой.
71) Основными типами боридов являются, по-видимому, для бериллия !!!!, а для магния — !!!!.. Наиболее характерны из них медно-красный Ве2В и черный MgB2. Бориды с относительно малым содержанием бора кислотами (или даже водой) разлагаются, с большим — не разлагаются.
72) Для бериллия довольно характерно образование с некоторыми другими металлами соединений (“бериллидов”), обычно содержащих относительно много атомов Be. Примерами могут служить !!!!.. Последнее из этих соединений находит использование в качестве источника нейтронов.
73) Производные одновалентных бериллия и магния не получены. В качестве промежуточных продуктов ионы Be+ и Mg+ образуются, по-видимому, при анодном окислении этих металлов. С равновесием по схеме!!! связана, вероятно, частичная (порядка одного атома на 100 молекул) растворимость магния в расплаве его хлорида.
Существование BeF и Be!!! при высоких температурах установлено для систем ВеГ2(г) + Ве(ж) Ы 2 ВеГ(г). Теплота образования газообразного BeF из элементовположительна (40 ккал/моль)а BeCl — отрицательна (-4 ккал/моль). Теоретические расчеты показывают, что галогениды ЭГ не должны устойчиво существовать при обычных условиях (так как их дисмутация на ЭГ2 и Э энергетически выгодна). Напротив, при очень высоких температурах одновалентное состояние становится устойчивее двухвалентного, как то видно из показанных на рис. результатов теоретического расчета равновесий термической диссоциации хлоридов магния.
74) Нульвалентное состояние Bе и Mg представлено их малоизученными дипиридильными производными — Э(Dipу)2. Они представляют собой тёмноокрашенные твердые вещества, растворы которых в тетрагидрофуране (при отсутствии даже следов кислорода) имеют зеленый (Be) или красный (Mg) цвет.

Рис. XII-6. Термическая
диссоциация магния.
заметным лишь немного выше 100, для MgCO3 — около 500 °С. CO2 MgO (MgO)
MgCO3
MgCO3 + H2O + CO2 = Mg(HCO3)2
CO2
Be (NH42CO3 M2[Be(CO3)2] K2[Mg(CO3)2]·4H2O KH[Mg(CO3)2]·4H2O Ca[Mg(CO3)2]. (NH4)2CO3
Mg(ClO4)2·6H2O Mg(ClO4)2
Be(ClO4)2·4H2O [Be(OH2)4](ClO4)2
Be(CH3COO)2 BeCl2
Mg(CH3COO)2·4H2O Mg(CH3COO)2 O®Mg
BeC2O4·3H2O BeSO4 Be[Be(C2O4)2]·6H2O BeC2O4 Na2[Be(C2O4)2]·H2O
47
33-74
По химическому характеру оба рассматриваемых элемента, в общем похожи друг на друга. Основные различия между ними связаны со значительным увеличением ионного радиуса при переходе от Be2+ (34 пм) к Mg2+(78 пм).
30
Третья группа периодической системы
Атомы элементов данной группы содержат во внешнем слое максимально по три электрона. Поэтому тенденция к дальнейшему присоединению электронов (с пополнением внешнего слоя до октета) не может быть для них характерна. Напротив, металлические свойства бора и его аналогов должны быть выражены сильнее, чем у соответствующих элементов четвёртой группы.
По аналогии с подгруппой титана можно ожидать, что элементы подгруппы скандия будут иметь тенденцию к отдаче не только двух электронов внешнего, но и лишнего против октета электрона следующего слоя, т. е. будут функционировать преимущественно как трёхвалентные металлы. С другой стороны, по аналогии с подгруппой германия можно ожидать, что Ga In и Tl будут способны проявлять в соединениях и более низкую валентность.
К своему ближайшему аналогу — алюминию — бор относится приблизительно так же, как углерод к кремнию. Сходство между обоими элементами ограничивается преимущественно их одинаковой валентностью и непосредственно обусловленными ею свойствами. По многим характеристикам бор существенно отличается от алюминия, и в целом его химия похожа скорее на химию кремния.
Бор
Он относится к распространённым: содержание его в земной коре составляет около 5·10-4 %. Скопления бора встречаются в виде кислородных соединений — борной кислоты (Н3ВО3), буры (Na2B4O7·10H2O), ашарита (MgHBO3) и ряда более сложных минералов. Ежегодная мировая добыча соединений бора исчисляется сотнями тысяч тонн.
Бура была известна алхимикам и упоминается ещё в сочинениях Гебера. Элементарный бор впервые получен в 1808 г. Природный элемент слагается их двух изотопов, относительное содержание которых подвержено небольшим колебаниям 10В (19,6-19,8 %) и 11В (80,4-80,2 %). Поэтому атомный вес его даётся с точностью ± 0,003.
В основном состоянии атом бора имеет внешнюю электронную оболочку 2s22p1 и одновалентен. Возбуждение его до трёхвалентного состояния (2s12p2) требует затраты 343 кДж/моль.
Наибольшие количества бора входят в состав буровых вод нефтяных месторождений и золы многих каменных углей. Наземные растения содержат 10-4-10-2 вес. % бора от сухого вещества (причём в злаках его меньше, а в корнеплодах больше). Животные организмы гораздо беднее бором. Внесение в почву соединений бора часто ведёт к существенному повышению урожайности культурных растений ( в частности льна и сахарной свёклы). Особенно сильно сказывается влияние бора на подзолистых почвах.
В свободном состоянии бор может быть получен из борной кислоты. Нагреванием её переводят в борный ангидрид (В2О3), который затем прокаливают с металлическим магнием. Реакция идёт по уравнению:
В2О3 + 3 Mg = 3 MgO + 2 B + 531 кДж.
После обработки продуктов реакции соляной кислотой (для удаления MgO) остаётся элементарный бор в виде тёмно-бурого порошка. Очень чистый бор бесцветен. Он имеет плотность 2,3 г/см3, плавится при 2075 и кипит при 3700 °С.
Весьма чистый (99,999 %) элементарный бор был получен восстановлением ВCl3 водородом при 1200 °С. Он может быть получен также термическим разложением паров ВВr3 на нагреваемой электрическим током до 1500 °С танталовой проволоке. Образующиеся очень мелкие кристаллы бора по твёрдости лишь немногим уступают алмазу. Они известны в четырёх различных кристаллических формах (имеющих сложное внутреннее строение), обладают металлическим блеском и при обычных условиях довольно плохо проводят электрический ток, но нагревание до 800 °С вызывает повышение электропроводности приблизительно в миллион раз (причём электронный характер низкотемпературной проводимости меняется при высоких температурах на дырочный). Теплота плавления бора оценивается в 22,6, теплота испарения — в 539, а теплота атомизации (при 25 °С) — в 560 кДж/моль. Помимо отдельных атомов пары бора частично содержат молекулы В2 энергия диссоциации которых оценивается в 276 кДж/моль.
Термическим разложением BI3 при 900 °С была получена аллотропная форма бора, имеющая красный цвет (вероятно от следов иода) и более простое строение кристаллической решётки. Выше 1500 °С она переходит в обычную форму.
В обычных условиях бор весьма инертен и взаимодействует лишь со фтором. Напротив, при высоких температурах он соединяется не только с кислородом, хлором и бромом, но и с серой, азотом и углеродом. При очень сильном прокаливании бор вытесняет соответствующие свободные элементы даже из таких устойчивых оксидов, как Р2О5, СО2 и SiO2, а также из оксидов многих металлов. В результате сплавления бора с некоторыми металлами образуются из бориды, например MgB2.
По отношению к воздуху и воде бор вполне устойчив. Взаимодействие его с водяным паром по схеме:
3 Н2О + 2 В = В2О3 + 3 Н2 + 548 кДж
идёт лишь при температуре красного каления. В кислотах, не являющихся окислителями, бор не растворяется. Концентрированная азотная кислота постепенно окисляет его до борной кислоты:
В + 3 HNO3 = H3BO3 + 3 NO2.
По отношению к обычно применяемым растворам щелочей бор устойчив. В своих соединениях он трёхвалентен.
Химическая активность бора зависит от степени его дробления. В явно кристаллическом состоянии он гораздо более инертен, чем в обычно получаемом мелко раздробленном (“аморфном”). Например, кристаллический бор устойчив по отношению к крепким растворам щелочей даже при кипячении, тогда как аморфный медленно реагирует с ними по схеме:
2 В + 2 NaOH + 2 H2O = 2 NaBO2 + 3 H2.
Даже расплавленные щёлочи более или менее быстро взаимодействуют с кристаллическим бором лишь в присутствии окислителей.
Подобно нитридам, карбидам и силицидам, некоторые из боридов по своему составу формально отвечают валентностям, известным для соответствующих элементов. Таковы, например, MnB, MnB2, CrB, CrB2, MoB2, WB2, VB, TiB. В других случаях это не соблюдается: примерами могут служить бориды общей формулы ЭВ2, где Э —Mg, V, Nb, Ta, Ti, Zr, Hf. Как правило, бориды образуются из элементов с выделением тепла, обладают высокой твёрдостью и хорошей электропроводностью. Многие из них отличаются очень высокими точками плавлению. Например, для ZrB2 и HfB2 они лежат соответственно при 3040 и 3250 °С. Кермет из борида циркония с металлическим хромом (как связкой) находит использование в ракетной технике. Устойчивость большинства боридов по отношению к кислотам довольно высока. Для типа ЭВ2 она возрастает по ряду MgB2 « VB22 2 2 2 2, причём MgB2 разлагается не только любыми кислотами, но и водой, а на TaB2 (т. пл. 3200 °С) не действует даже кипящая царская водка.
Наиболее характерны для бора кислородные соединения. При нагревании на воздухе до 700 °С он сгорает красноватым пламенем с образованием борного ангидрида по схеме:
4 В + 3 О2 = 2 В2О3.
Практически В2О3 удобнее получать прокаливанием Н3ВО3. Борный ангидрид представляет собой бесцветную стекловидную массу.
Теплота образования кристаллической формы В2О3 из элементов равна 1275, а обычной стеклообразной — 1254 кДж/моль. Последняя обладает высокой твёрдостью, но начинает размягчаться уже выше 200 °С и не имеет четкой температуры плавления. Кристаллическая форма плавится при 450 (теплота плавления 25 кДж/моль) и кипит при 2200 °С. Пар борного ангидрида состоит из термически устойчивых молекул В2О3, строение которых выражается формулой О=В-О-В=О с плоской угловой структурой.
Расплавленный В2О3 при высоких температурах хорошо растворяет оксиды многих элементов. Хуже других растворяются в нём ВеО (0,2 вес. %), TiO2 (0,6), SnO2 (0,8) Al2O3 (1).
При нагревании смеси В2О3 с элементарным бором выше 1000 °С в парах преобладают термически устойчивые линейные молекулы О=В-В=О. Прочность связи В-В в них превышает 420 кДж/моль, и диссоциация по схеме MnB = 2 ВО практически не наблюдается. Охлаждение паров сопровождается дисмутацией оксида бора (II) по схеме: 3 В2О2 = 2 В2О3 + 2 В с образованием коричневой смеси обоих продуктов распада.
Однако “замораживанием” системы путём её быстрого охлаждения ниже 300 °С может быть получен белый твёрдый полимер (В2О2)х. Он не имеет определённой точки плавления, рентгеноаморфен и весьма реакционноспособен, а при нагревании выше 300 °С дисмутирует по приведённому выше уравнению.
Под давлением около 60 тыс. атм и при температуре порядка 1500 °С взаимодействие В2О3 с элементарным бором идёт по схеме: В2О3 + 4 В = 3 В2О. Этот низший оксид бора имеет слоистую структуру типа графита. Сообщалось также о получении оксидов состава В6О и В7О.
На воздухе В2О3 притягивает влагу, а в воде растворяется с образованием борной кислоты по схеме:
В2О3 + 3 Н2О = 2 Н3ВО3.
Борная, (точнее, ортоборная) кислота представляет собой бесцветные кристаллы, сравнительно малорастворимые в воде. При нагревании она теряет воду и переходит сначала в метаборную кислоту (НВО2), а затем в борный ангидрид. Растворение этих веществ в воде сопровождается обратным переходом в Н3ВО3.
Пространственная структура иона ВО33- отвечает плоскому равностороннему треугольнику [d(BO) = 136 пм]. Имеющие вид блестящих чешуек пластинчатые кристаллы Н3ВО3 строится сочетанием таких ионов друг с другом посредством водородных связей, причём образуются слагающиеся из правильных шестиугольников плоскости с расстоянием 318 пм между ними. Так как эти плоскости лишь слабо связаны друг с другом (за счёт межмолекулярных сил), кристаллы легко делятся на отдельные слои.
Борная кислота (К1 = 6·10-10, К2 = 5·10-13, К3 = 4·10-14) окрашивает пламя в характерный зелёный цвет (обусловленный, по-видимому, электронными переходами в молекуле В2О3). Она несколько летуча с водяным паром и содержится в воде некоторых горячих источников. Её насыщенный водный раствор содержит около 4,5 % Н3ВО3 при обычных условиях и около 28 % при 100 °С. Весьма вероятно, что в водном растворе молекула борной кислоты образует донорно-акцепторную связь с одной молекулой воды и её первичная диссоциация идёт по схеме:
Н2ОВ(ОН)3Ы Н· + [B(OH)4]’.
Это подтверждается, в частности, рентгеновским анализом соли состава NaBO2·4H2O, структура которой оказалась отвечающей формуле Na[B(OH)4]·2H2O с тетраэдрическим окружением атома бора гидроксильными группами.
Помимо воды, борная кислота хорошо растворима с спирте, хуже растворима она в ацетоне (5 г/л) и почти нерастворима в эфире (0,08 г/л). Взаимодействие с глицерином усиливает кислотные свойства Н3ВО3. Они возрастают также по мере повышения концентрации самой кислоты. Так, при переходе от 0,03 к 0,75 М водному раствору рН изменяется от 5,3 до 3,7, что связано с частичным образованием полиборных кислот (предположительно, три- и гексаборной).
Борная кислота непосредственно используется при эмалировании железных сосудов (вводится в состав эмалей) и в медицине (как дезинфицирующее средство), а также служит обычным исходным продуктом для получения остальных соединений бора. Кислота эта является хорошим дезинфицирующим и консервирующим средством, однако применение её в пищевой промышленности недопустимо, так как она вызывает расстройство пищеварения.
Переход Н3ВО3 в НВО2 начинается около 100 °С. Метаборная кислота известна в трёх различных кристаллических формах с температурами плавления 176, 201 и 236 °С. Наиболее тугоплавкая форма растворяется в воде (с переходом в Н3ВО3) значительнее медленнее двух других.
При взаимодействии борной кислоты со спиртами в присутствии концентрированной H2SO4 (для связывания воды) легко идёт образование эфиров по схеме, например:
3 СН3ОН + Н3ВО3 = 3 Н2О + В(ОСН3)3.
Пары образующегося борнометилового эфира (т. пл. -29, т. кип. 69 °С) при поджигании горят бледно-зелёным пламенем, чем пользуется как качественной реакцией на бораты. На том же принципе может быть основано количественное выделение борной кислоты (с одновременным переводом боратов в сульфаты).
Диссоциация гидроксида В(ОН)3 идёт по кислотному типу. Однако борная кислота очень слаба и поэтому из растворов её солей легко выделяется большинством других кислот. Соли её (бораты) производятся обычно от различных полиборных кислот общей формулы nB2O3·mH2O, чаще всего — тетраборной (n = 2, m = 1). Последняя является кислотой значительно более сильной, чем ортоборная.
Соли Н2В4О7 образуются при нейтрализации Н3ВО3 щелочами, например, по схеме:
2 NaOH + 4 H3BO3 = Na2B4O7 + 7 H2O.
Избытком щёлочи они могут быть переведены в метабораты:
2 NaOH + Na2B4O7 = 4 NaBO2 + H2O
C другой стороны, при действии на тетрабораты (или метабораты) сильных кислот выделяется свободная ортоборная кислота:
Na2B4O7 + H2SO4 + 5 H2O = Na2SO4 + 4 H3BO3
В воде из боратов растворимы только соли наиболее активных одновалентных металлов. Вследствие гидролиза растворы их показывают сильнощелочную реакцию.
Важнейшим для практики боратом является натриевая соль тетраборной кислоты — бура. Она выделяется из раствора в виде бесцветных кристаллов состава Na2B4O7·10H2O, выветривающихся на воздухе и при обычных условиях малорастворимых в воде.
Так как безводные бораты чрезвычайно устойчивы по отношению к нагреванию, при высоких температурах борная кислота выделяет большинство других кислот из их солей. В этом отношении (как и по своей слабости) она похожа на кремневую кислоту.
Метабораты щелочных металлов — LiBO2 (т. пл. 833 °С), NaBO2 (т. пл. 966) и КВО2 (т. пл. 947) — весьма термически устойчивы и при достаточном нагревании испаряются без разложения. В парах молекулы мономерны. В твёрдом состоянии эти соли тримерны, причём анион В3О63- имеет показанное на рис. плоское циклическое строение. При такой же тройной координации атомов бора кристалл Са(ВО2)2 содержит полимерные цепные анионы. Сходное с метаборатом кальция строение имеет Pb(BO2)2 (т. пл. 868 °С). Эта нерастворимая в воде и негигроскопичная соль находит использование при изготовлении защитных экранов для ядерных реакторов.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() О О
О О
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() НО В В О В В ОН
НО В В О В В ОН
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() О О
О О
Рис. 1. Строение иона В3О63-. Рис. 2. Структура Н2В4О7.
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Рис. 3. Строение цепи (ВО2)nn-.
Выделяемые из растворов метабораты обычно содержат кристаллизационную воду. Возможно, что на самого деле они являются кислыми ортоборатами. В частности, для Са(ВО2)2·2Н2О (правильнее структура Са(НВО3)2.
Неизвестной в свободном состоянии тетраборной кислоте (К1=2·10-4, К2=2·10-5) может быть придана структурная формула (рис. 2). Для её натриевой соли, помимо обычной буры, характерен также кристаллогидрат Na2B4O7·5H2O (“ювелирная бура”), осаждающийся из растворов выше 56 °С и на воздухе не выветривающийся. Насыщенный водный раствор буры содержит около 2,5 % тетрабората натрия при обычных условиях (рН = 9,3) и около 33 % при 100 °С. Бура растворима также в спирте и глицерине. Безводный Na2B4O7 (т. пл. 741 °С) может быть получен нагреванием буры выше 400 °С (но образующаяся первоначально аморфная фаза закристаллизовывается лишь при 675 °С).
Бура потребляется рядом различных производств (стекольным, керамическим, кожевенным и др.). Она находит также медицинское использование (как дезинфицирующее средство) и входит в состав некоторых стиральных порошков.
Из солей различных полиборных кислот многие встречаются в природе и служат исходными продуктами для получения соединений бора. Таков, например, минерал гидроборацит — СаМgB6O11·6H2O, производящийся от гексаборной кислоты (n = 3, m = 2). Наряду с солями полиборных кислот, являющихся изополикислотами бора, известны также производные некоторых его гетерополикислот, например H9[B(W2O7)6]. Последняя отвечает неизвестному в свободном состоянии гидрату H9BO6 (т. е. B2O3·9H2O).
Перевод солей других кислот в бораты путём их сплавления с избытком H3BO3 происходит при различных температурах, например, для KNO3 при 500, для KCl при 800 и для K2SO4 при 1000 °С. Бораты образуются также при сплавлении солей или оксидов с бурой, например, по схеме:
Na2B4O7 + CoO = 2 NaBO2 + Co(BO2)2.
Так как бораты некоторых металлов характерно окрашены, растворы их в расплавленной буре образуют цветные стёкла (например, синее для кобальта или зелёные для хрома). Этим пользуются иногда в аналитический химии для открытия таких металлов. Обычно реакцию проводят в ушке платиновой проволочки, причём получается окрашенная капля борного стекла (“перл буры”).
Помимо буры и борной кислоты некоторое практическое значение имеют также соли надборных кислот (пербораты), образующиеся путём замены атомов кислорода в борате на пероксидные группы -О-О-. Свободные надборные кислоты не выделены, но в растворах они существуют. На это указывает заметное повышение кислотности растворов Н3ВО3 при добавлении к ним Н2О2 (для К1 даётся значение 2·10-8).
Чаще всего встречается в практике перборат состава NaBO3·4H2O может быть получен действием H2O2 на раствор метабората натрия и представляет собой бесцветные кристаллы, сравнительно малорастворимые в воде (около 25 г/л при обычных условиях). По данным рентгеноструктурного анализа, строение этой соли отвечает формуле Na2[(HO)2B(O2)2B(OH)2]·6H2O, т. е. она является производным истинной надборной кислоты. Вместе с тем, в отличие от солей других надкислот, она не выделяет иод из раствора KI. На этом основании её обычно трактовали как метаборат натрия, содержащий кристаллизационный пероксид водорода, т. е. NaBO21·H2O2·3H2O. По-видимому, в растворе этой соли имеет место сильно смещённое вправо гидролитическое равновесие по схемам: Na2[(OH)2B(O2)2B(OH)2] + 4 H2O Ы 2 Na[(HO)2BO2] + 4 H2O Ы 2 Na[B(OH)4] + 2 H2O2, чем и обусловлено отсутствие выделения иода. Обезвоживанием тригидрата могут быть получены NaBO2·H2O2·2H2O и NaBO2·H2O2. Все три соли устойчивы при хранении без доступа воздуха. Они применяются главным образом для отбелки различных материалов и часто вводятся в состав стиральных порошков.
Из растворов соответствующих метаборатов в 30%-ном H2O2 могут быть выделены (путём обезвоживания над P2O5 под уменьшенным давлением) бесцветные кристаллические соли состава LiBO4·H2O, ЭBO4·1/2H2O (где Э — Rb, Cs) и KBO5·H2O. Соединения эти являются истинными перборатами, причём они производятся от ортонадборной кислоты(т. е. отвечает формуле KH2BO6). Были получены также безводные пербораты калия и аммония — KBO3, KBO4, NH4BO3 и NH4BO3·NH4BO4.
Хотя основные свойства для B(OH)3 нехарактерны, однако некоторые солеобразные соединения бора известны. Его фосфорное производное получается в виде белого порошка при взаимодействии растворов B(OH)3 и HPO3 в концентрированной CH3COOH. Соль эта имеет состав (BO)PO3 и производится не от иона В3+, а от одновалентного радикала борила — BO+, аналогичного титанилу, цирконилу и т. п. Удобнее получать метафосфат борила прокаливанием до 800 °С смеси борной кислоты с фосфатом аммония. Известен и аналогичный фосфату по свойствам арсенат борила — (BO)AsO3. Встречающимся в природе представителем соединений этого типа может служить минерал датолит —Ca2(BO)2(SiO3)2(OH)2. По реакции:
BCl3 + 3 ClNO2 = 3 Cl2 + 2 NO + (BO)NO3
был получен устойчивый при низких температурах нитрат борила.
Наиболее давно известной солью непосредственно трёхвалентного бора является его ацетат — B(CH3COO)3 (т. пл. 149 °С). От него производятся комплексы M[B(CH3COO)4] (где M — Cs, Rb, K, Tl). Взаимодействием BCl3 с безводной HClO4 был получен кристаллических перхлорат бора — B(ClO4)3. Соль эта сама по себе устойчива лишь ниже -5 °С, но может быть стабилизирована присоединением триметиламина. Малоустойчивые смешанные хлористо-хлорнокислые соли — BCl2(ClO4) и BCl(ClO4)2 — имеют тенденцию к саморазложению на BCl3 и B(ClO4)3.
Нитрат трёхвалентного бора известен в виде комплексной соли тетраметиламмония — [N(CH3)4][B(NO3)4]. Это белое твёрдое вещество было получено взаимодействием [N(CH3)4][BCl4] с N2O4 при температуре -78 °С, но в отсутствии влаги оно устойчиво и при обычной температуре.
Гидросульфат бора — B(HSO4)3 — образуется при взаимодействии B(OH)3 с безводной серной кислотой. Удобнее его получать по реакции:
BCl3 + 3 H2SO4 = 3 HCl + B(HSO4)3.
Гидросульфат бора представляет собой гигроскопичный белый порошок ещё не плавящийся при 240 °С. С безводной H2SO4 он образует комплексную кислоту H[B(HSO4)4], которая также была выделена в твёрдом состоянии. Получены и некоторые соли этой кислоты (с Na, K, NH4, Sr). Гораздо шире представлены соли серноборной кислоты H[B(SO4)2], известные для многих одновалентных и двухвалентных металлов. Образуются они при нагревании H3BO3 и соответствующих сульфатов в безводной H2SO4.
Бесцветный сульфид бора — B2S3 образуется при нагревании бора выше 600 °С в парах серы:
2 B + 3 S = B2S3 + 251 кДж.
Более удобным способом его получения является прокаливание аморфного бора в токе сухого H2S. Сульфид бора может быть перекристаллизован из раствора в PCl3 и получен в виде белых игл, плавящихся при 310 °С и в токе H2S легко возгоняющихся. Молекула S=B-S-B=S имеет плоское угловое строение РBSB = 96°. С галогенидами бора и аммиаком сульфид бора образует кристаллические продукты присоединения (в частности, жёлтый B2S3·6NH3), а водой полностью разлагается на борную кислоту и H2S. Был описан и другой сульфид бора — жёлтый B2S5 (но в его индивидуальной природе нет уверенности). Получен также аналогичный сульфиду жёлтый селенид бора — B2Se3.
Сплавлением B2S3 с фосфором и серой получено (в двух модификациях — бесцветной и коричневой) кристаллическое вещество состава BPS4, которое можно рассматривать как сернистый аналог метафосфата борила. Действием сероводорода на BBr3 было получено в виде белых игл сернистой производное бора, отвечающее по составу тиометаборной кислоте. Кристаллы этого соединения образованы тримерными молекулами (HBS2)3 имеющими циклическую структуру (из групп BSH и атомов S). Оно отщепляет H2S уже при обычной температуре, водой тотчас гидролизуется, а в бензоле растворяется без изменения. Отвечающей этой кислоте белые тиометабораты — NaBS2 (т. пл. 580 °С) и KBS2 (т. пл. 550 °С) — на воздухе тотчас гидролизуются. Известны также тиопербораты — жёлтые MBS3 и бледно-желтые M2B2S5 (где M — Na, K).
С азотом бор соединяется только выше 1200 °С. Нитрид бора BN может быть получен также прокаливанием бора (или B2O3) в атмосфере аммиака. Он образуется из элементов с выделением тепла (250 кДж/моль) и представляет собой белый, похожий на тальк порошок, плавящийся лишь около 3000 °С (под давлением азота). Плотность частиц этого порошка равна 2,3 г/см3, а по смазочным свойствам он превосходит и графит и MoS2. В спрессованном состоянии нитрид бора обладает полупроводниковыми свойствами (с шириной запрещённой зоны около 3Э7 эВ), а при наличии небольших примесей С и B2O3 сильно фосфоресцирует после предварительного освещения. Выше 1000 °С он начинает разлагаться на элементы (при 1200 °С давление азота составляет 0,3 мм рт. ст.).
При обычных условиях нитрид бора химически инертен — не реагирует с кислородом или хлором, кислотами или щелочами. Однако в токе фтора он самовоспламеняется и сгорает по уравнению:
2 BN + 3 F2 = 2 BF3 + N2,
а фтористоводородная кислота разлагает его с образованием NH4BF4. Под действием горячих растворов щелочей (или паров воды при температуре красного каления) BN разлагается с выделением аммиака. Кислород и хлор начинают действовать на него лишь выше 700 °С.
По кристаллической структуре обычная форма BN сходна с графитом [d(BN) = 145 пм], но шестиугольники располагаются точно друг над другом с чередованием атомов B и N в соседних слоях, расстояние между которыми составляют 333 пм. В отличие от графита отдельные кристаллики BN прозрачны. По вопросу о возможности образования им продуктов внедрения (аналогичным графитидам) имеются противоречивые данные, но аддукты щелочных металлов существуют. Были получены также смешанные нитриды бора — Li3ВN2 и Э3(BN2)2, где Э — Ca, Ba. Водой они разлагаются.
При давлении выше 62 тыс. атм и температурах выше 1350 °С обычная графитоподобная структура BN изменяется на алмазоподобную, в которой половина атомов С замещена на атомы В, а другая половина — на атомы N с расстоянием d(BN) = 157 пм. Хорошим катализатором такого превращения являются щелочные и щелочноземельные металлы. Как и в случае перехода графит ® алмаз, оно сопровождается резким изменением свойств нитрида бора.
Алмазоподобная форма этого вещества — “боразон”, или “эльбор”, — получаются обычно в виде мелких кристаллов различной окраски одинаковой с алмазом плотностью и твёрдостью, но сильно превосходит алмаз по термостойкости (до 2000 °С) и ударной прочности. Подобно алмазу, он является электроизолятором, но некоторыми примесями может быть переведён в полупроводниковое состояние как n-типа (S), так и p-типа (Be). Химическая стойкость боразона значительно выше, чем обычной формы нитрида бора.
Важным достоинством эльбора является устойчивость оснащённого им режущего инструмента (резцов, свёрл и др.) при скоростной обработке стали и чугуна. Алмаз для этого мало пригоден, так как контакт с раскалённым железом сильно ускоряет его графитизацию.
Из четырёх валентных связей каждого атома боразона три являются обычными, а четвёртая — донорно-акцепторной B®N, что даёт формальные заряды N+ и B-. Между тем оценка фактических эффективных зарядов приводит к обратным по знакам значениям +0,8 для В и -0,8 для N. Последние имеют порядок величин, характерный для атомов в кристаллах типичных солей (например, NaCl). Таким образом, валентную связь в боразоне можно с полным основанием назвать ковалентно-ионной.
С фосфором бор соединяется только около 1000 °С, образуя коричневый фосфид — BP. Последний, подобно боразону, имеет алмазоподобную структуру и высокую твёрдость (большую, чем у кварца). Он устойчив по отношению к нагреванию (переходит в серый B13P2 лишь выше 1180 °С) и в кристаллическом состоянии при обычных условиях весьма химически инертен. Фосфид бора обладает свойствами полупроводника с большой шириной запрещённой зоны (4,5 эВ). Известен и похожий по свойствам на фосфид арсенид бора — BAs.
Карбид бора B4C образуется в виде чёрных блестящих кристаллов при прокаливании смеси бора (или B2O3) с углём в электрической печи. Кристаллы эти слагаются по типу решётки NaCl из линейных групп C3 и группировок В12, в которых атомы бора располагаются по углам икосаэдра. Карбид бора (теплота образования из элементов 71 кДж/моль) имеет плотность 2,5 г/см3, отличается тугоплавкостью (т. пл. 2360 °С), довольно хорошей для неметалла электропроводностью (примерно 0,001 от электропроводности ртути), чрезвычайной твёрдостью (близкой к алмазу) и высокой устойчивостью по отношению к различным химическим воздействиям. Например, ниже 1000 °С на него почти не действуют пи хлор, ни кислород (а взаимодействие с водяным паром при 900 °С идёт по уравнению:
В4С + 6 Н2О = 2 В2О3 + С + 6 Н2 и затем В2О3 + Н2О = 2 НВО2.
Карбид бора находит использование при выработке и обработке различных твёрдых сплавов, а также в атомной промышленности (для улавливания нейтронов). Из силицидов бора известны B3Si и B6Si.
Галогениды бора общей формулы ВГ3 могут быть получены взаимодействием элементов при обычных условиях (F), при 400 (Сl), 700 (Br) или 900 °С (I).
Для получения BF3 более применим другой метод: нагревание смеси B2O3 и CaF2 с концентрированной серной кислотой. Реакция при этих условиях идёт по суммарному уравнению:
B2O3 + 3 CaF2 + 3 H2SO4 = 2 BF3 + 3 CaSO4 + 3 H2O.
Чистый сухой BF3 удобно получать термическим разложением Ва(BF4)2, быстро протекающим уже при 500 °С.
Они представляют собой бесцветные вещества, дымящие во влажном воздухе. Строение молекул галогенидов ВГ3 отвечает плоскому треугольнику с атомам В в центре.
BF3 | BCl3 | BBr3 | BI3 | |
| Теплота образования, кДж/моль | 1137 | 426 | 238 | 38 |
| d(BГ), пм | 131 | 174 | 189 | 210 |
Энергия связи В-Г, кДж/моль | 644 | 443 | 376 | 284 |
Температура плавления, °С | -128 | -107 | -46 | +50 |
Температура кипения, °С | -100 | +13 | 90 | 210 |
Критическая температура, °С | -12 | 179 | 300 |
Фторид BF3 и хлорид ВСI3 при обычных условиях газообразны, BBr3 — жидкость и BI3 — твёрдое тело. Водой галогениды бора (кроме BF3) разлагаются по схеме:
ВГ3 + 3 Н2О = В(ОН)3 + 3 НГ.
В отличие от своих аналогов ВF3 гидролизуется незначительно.
Устойчивость галогенидов бора уменьшается от F®I: если BF3 чрезвычайно термически стоек, то BI3 под действием света разлагается уже при обычных условиях. Пары его действуют на кварц. Взаимодействие при высоких температурах BCl3 и BВr3 с оксидами некоторых металлов могут быть получены их безводные хлориды или бромиды. Для эффективного заряда атома бора в BF3 даётся значение +1,42 (по другим данным +1,29), а для энергий последовательного отрыва атомов фтора — значения 706, 493, 727 кДж/моль. Фторид бора является хорошим катализатором некоторых органических реакций.
Частично образующиеся при взаимодействии различных BГ3 смешанные галогениды бора имеют сильно выраженную тенденцию к симметризации и в индивидуальном состоянии неустойчивы. То же относится и к газообразным при обычных условиях гидрогалогенидам бора — HBF2 и HBCl2. Первое из этих соединений d(BF) = 131, d(BH) = 119 пм способно присоединять этилен с образованием C2H5BF2.
Интересно протекает взаимодействие галогенидов бора с галогеноводородами. В газообразной системе
BX3 + 3 HY Ы BY3 + 3 HX
равновесие быстро смещается вправо, если галогенид Y стоит в периодической системе выше галоида X, и влево, если X стоит выше Y. Например, из BI3 и HВr легко образуются BВr3 и HI, тогда как обратный перевод осуществляется лишь при 300-400 °С и в незначительной степени.
Фторид бора умеренно растворим в бензоле (около 7:10 по объёму) и очень хорошо в воде (до 1000:1 по объёму при 0 °С). Как и в случае кремния фторид относится к воде иначе, чем другие галогениды бора. Он не подвергается полному гидролизу, а реагирует, в основном, с образованием гидроксофтороборной кислоты по схеме:
H2O + BF3Ы H[HOBF3].
Её составу отвечает моногидрат фторида бора — H2O·BF3 (т. пл. 6 °С). Как одноосновная, она является очень сильной, но с основаниями может реагировать и в качестве гораздо менее сильной двухосновной оксофтороборной кислоты — H2[OBF3]. Например, известны соли состава K[HOBF3] и Ba[OBF3] (а также аналогичное первой из этих солей аминопроизводное — K[H2NBF3].
Кристаллогидрат BF3·2H2O (т. пл. 6 °С) представляет собой оксониевую соль гидроксофтороборной кислоты — (H3O)[HOBF3]. Интересно, что в его ионе [HOBF3]- средняя длина связи B-F (137 пм) промежуточна между длиной аналогичной связи в BF3 (131) и BF4- (143), а длина связи O-B (156) значительно больше её обычного среднего значения (147 пм). Это указывает как будто на более активное взаимодействие бора с фтором, чем с гидроксилом. Однако возможна и другая трактовка структуры рассматриваемого соединения — как комплексов H2O·BF3 и молекул H2O, соединяющих эти комплексы друг с другом водородными связями.
Процесс частичного гидролиза гидроксофтороборной кислоты по схеме:
H2O + H[HOBF3] Ы HF + H[(HO)2BF2]
— известна только в жидком состоянии H[(HO)2BF2] — является не свободной кислотой, а тримерной оксониевой солью (H3O)3[O3B3F6]3 с шестичленным циклическим (из атомов кислорода и групп BF2-) строением аниона. Были получены и некоторые аналогичные металлические (Na, K) производные. Например, полученный взаимодействием KF с борной кислотой кристаллический K[(HO)3BF], по-видимому, мономерен.
Строение шестичленного цикла (из атомов кислорода и групп BГ) характерно для оксогалогенидов бора — O3B3Г3 (где Г — F, Cl, Br), образующихся в виде возгонов при взаимодействии галогенидов BГ3 с нагретым выше 200 °С борным ангидридом. Ниже этой температуры они распадаются на исходные вещества. Для фторида около 1000 °С под уменьшенным давлением установлено наличие диссоциации в парах по схеме:
O3B3F3Ы 3 OBF
Такая диссоциация ещё более характерна для хлорида.
Были получены и аналогичные по строению тиогалогениды бора — S3B3Г3 (где Г — Cl, Br). В отличии от оксогалогенидов, они устойчивы лишь при низких температурах (ниже 20 °С.
Известны, но ещё плохо изучены, и некоторые аналогичные галогениды BГ3 производные бора. Длительным контактом BСl3 с AgCN был получен цианид бора [B(CN)3], взаимодействием BСl3 с KCNS в жидкой SO2 — его роданид [B(NCS)3], из BСl3 и NaCCH — его гидроацетилид [B(CCH)3], а из B2H6 и HN3 — его азид (B(N3)3. Описаны также некоторые смешанные производственные [например, (ВГ2N3)3, где (Г — Cl, Br) и двойные соединения — Li[В(NCS)4] (в виде эфирата) и M[B(N3)4] (где M — Li, Na). Все перечисленные вещества бесцветны, при обычных условиях твёрды и малоустойчивы. Азидные производные взрывчаты. Наличием прямой валентной связи бора с марганцем интересно неустойчивое на воздухе соединение состава R2BMn(CO)4PR3, где R — C6H5.
Кроме основного типа ВГ3 для бора известны низшие галогениды, содержащие в своей структуре связи В-В. Как правило, соединения эти малоустойчивы. Важнейшим из них является дибор террахлорид, получающийся по схеме:
2 BСl3 + 2 Hg = Hg2Cl2 + B2Cl4
пропусканием ВСl3 под давлением около 1 мм рт. ст. сквозь ртутную электрическую дугу. Образующийся B2Сl4 представляет собой бесцветную жидкость (т. пл. -93 °С), медленно разлагающуюся на BСl3 и (BСl)n уже выше 0 °С. Молекулы B2Cl4 имеет плоскую структуру, но в газообразном и жидком состоянии группы BСl2 располагаются перпендикулярно друг другу.
Даже при низких температурах дибор тетрахлорид энергично взаимодействует с кислородом, хлором и бромом (но не взаимодействует с серой и иодом). Водородом он разлагается в основном по схеме:
3 B2Cl4 + 3 H2 = 4 BCl3 + B2H6.
Первой стадией всех этих реакций B2Cl4 является, вероятно, присоединение им соответствующих молекул с разрывом связи B-B и последующей симметризацией образовавшихся смешанных производных. С аммиаком идёт реакция замещения по схеме:
B2Cl4 + 6 NH3 = 4 NH4Cl + B2(NH)2,
а с гидразином — по схеме:
B2Cl4 + 5 N2H4 = 4 N2H5Cl + B2N2.
Как амидное, так и нидридное производные представляют собой белые твёрдые вещества и являются полимерами. Последнее соединение отличается по свойствам от обычного нитрида бора и слагается из структурных элементов типа:
—B—B—N—N—. В качестве устойчивого мономерного соединения со
Ѕ Ѕ Ѕ Ѕ
связью B-B следует отметить [(CH3)2N]2B-B[N(CH3)2]. Вещество это в сухом воздухе выдерживает нагревание до 200 °С.
Для B2Cl4 известны и продукты присоединения многих веществ. Примером может служить белое, твёрдое и довольно термически устойчивое производное пиридина (C5H5N)2B2Cl4. Интересна протекающая в жидком хлористом водороде реакция по уравнению
2 [N(CH3)4]Cl + B2Cl4 = [N(CH3)4][B2Cl6],
результатом которой является осаждение белой соли тетраметиламмония и аниона [B2Cl6]2-.
В обоих приведённых выше случаях связь B-B не разрывалась. Напротив, присоединение этилена сопровождается разрывом этой связи с образованием Cl2BCH2CH2BCl2 (т. пл. -28 °С). Интересно, что в плоской (кроме атомов водорода) структуре рассматриваемой молекулы связь C-C имеет длину не 154, а 146 пм, обычно характерную для неё при соседстве двух двойных связей.
Взаимодействие B2Cl4 с водой идёт при обычных условиях по уравнению:
B2Cl4 + 4 H2O = 4 HСl + B2(OH)4
Выше 90 °С начинает играть роль вторичная реакция:
B2(OH)4 + 2 H2O = H2 + 2 B(OH)3
Отвечающая формуле (HO)2B-B(OH)2 или H4B2O4 кислота представляет собой белое кристаллическое вещество, хорошо растворимое в воде (и спирте). По силе она сравнима с ортоборной, но отличается от неё резко выраженной восстановительной активностью. Так, реакция по схеме:
H4B2O4 + O2 + H2O = 2 H3BO3
в щелочной среде заканчивается за несколько минут. Из-за этого, вероятно, до сих пор не получены соли H4B2O6. Последняя способна также к дисмутации по схеме:
3 B2(OH)4 = 4 B(OH)3 + 2 B.
При нагревании в вакууме H4B2O4 медленно теряют воду с образованием (B2O2)x. Полученный таким путём белый полимер монооксида бора менее реакционноспособен, чем образующийся при “замораживании” пара. С водой он даёт смесь B2(OH)4 и B(OH)3 относительное содержание которых зависит от условий взаимодействия.
Известны и более “ненасыщенные” кислоты бора — H6B2O2 и H4B2O2 (т. е. HOBH-HBOH). Первая из них (вероятно, в действительности HOBH2) образуется при обработке борида магния водой, а соли обеих кислот — при его взаимодействии с растворами щелочей разных концентраций. Из продуктов гидролиза борида магния была выделена и аммонийная соль “субтетраборной” кислоты — H2B4O6, строение которой подобно тетраборной, но с прямой связью между двумя центральными атомами бора. Термическим разложением этой соли по реакции:
(NH4)2B4O6 = 2 NH3 + B4O5 + H2O
был получен оксид бора B4O5, имеющий полимерный характер. Все эти “субборные” кислоты и их производные ещё плохо изучены.
Получаемая по схеме:
MgB2 + 4 H2O = Mg(OH)2Ї + 2 HOBH2
и затем (при прокаливании):
2 HOBH2 = 3 H2 + B2O2
монооксид бора может быть использован для синтеза B2Cl4, Дело в том, что при температурах около 250 °С реакция по схеме:
4 BСl3 + 3 B2O2 = 2 B2O3 + 3 B2Cl4
идёт с довольно хорошим выходом дибор тетрахлорида. Так как последний является большим исходным продуктом для получения многих других соединений бора, содержащих в своём составе связи B-B, получение его самого наиболее простым путём весьма желательно.
Взаимодействием по схеме:
4 SbF3 + 3 B2Cl4 = 3 B2F4 + 4 SbCl3
при низких температурах может быть получен дибор тетрафторид (т. пл. -56 °С, т. кип. -34 °С). Молекула его характеризуется следующими структурными параметрами: d(BF) = 132, d(BB) = 167 пм, РFBF = 120 °С. Энергия связи BB равна 431 кДж/моль, а вращение по ней почти свободно.
Термическая устойчивость B2F4 довольно высока — даже при 100 °С он разлагается [на BF3 и (BF)x] лишь медленно. Его химические свойства, в общем, подобны свойствам B2Cl4, но менее изучены. Интересно, что с SO2 дибор тетрафторид не реагирует, а с монооксидом ртути уже при низких температурах идёт реакция по уравнению:
3 B2F4 + 3 HgO = B2O3 + 4 BF3 + 3 Hg.
Ещё менее изучен бесцветный дибор тетрабромид (т. пл. 1 °С), который может быть получен по реакции:
3 B2Cl4 + 4 BВr3 = 4 BСl3 + 3 B2Br4.
Был получен и твёрдый при обычных условиях бледно-жёлтый B2I4.
Из продуктов термического разложения B2Cl4, помимо BСl3 и (BСl)x, могут быть в небольших количествах выделены два индивидуально охарактеризованных твёрдых субхлорида — бледно-жёлтый, довольно летучий B4Cl4 (т. пл. 95 °С) и красный B8Cl8. Они обладают высокой реакционной способностью (например, B4Cl4 на воздухе самовоспламеняется).
С позиции обычной теории валентности строение молекулы B4Cl4 должно было бы отвечать квадрату, образованному группами BСl. Однако результаты проведённого рентгеноструктурного анализа истолковываются в пользу тетраэдрического расположения атомов хлора d(BСl) = 170, d(BB) = 171 пм. Если это так, то каждый его атом должен осуществлять не три, а четыре ковалентные связи, на что у него не хватает внешних электронов, т. е. молекула является электронодефицитной. Предполагается, что связи B-Cl нормальные ковалентные, а остальные 8 электронов четырёх атомов бора попарно занимают четыре связывающие молекулярные орбитали тетраэдра. Приблизительно так же обстоит дело и с молекулой B8Cl8.
Субгалогениды бора состава (BГ)x представляет собой твёрдые вещества белого (F), жёлтого (Cl), красного (Br) или чёрного цвета (I). Отмечалось также существование красного B12Cl11 (т. пл. 115 °С) и не возгоняется до 350 °С и светло-жёлтого B9Cl9. Все эти вещества ещё очень мало изучены.
Для галогенидов бора весьма характерны реакции присоединения к ним молекул различных других веществ, в частности многих органических соединений. Наибольшее значение из таких производных имеет продукт присоединения HF к BF3 — комплексная тетрафтороборная кислота H[BF4]. Сама она устойчива только в растворе, причём её кислотные свойства выражены гораздо сильнее, чем у HF. Большинство солей HBF4(фторборатов) бесцветно и хорошо растворимо в воде.
Растворимость BF3 в жидком фтористом водороде невелика (порядка 0,5 мол % при обычных условиях) и друг с другом они химически не взаимодействуют. Напротив, в присутствии вещества, способного связывать H+ (например, воды) идёт реакция по схеме:
F- + BF3Ы BF4-.
Образующийся комплексный ион [BF4]- представляет собой правильный тетраэдр с d(FB) = 143 пм (т. е. значительно большим, чем в ВF3). Многие фторбораты хорошо кристаллизуются и выдерживают довольно сильное нагревание (например, КBF4 плавится при 530 °С без разложения). По растворимости они похожи на перхлораты: относительно малорастворимы производные К, Rb и Cs (порядка 1:200 по массе), а также некоторых объёмистых комплексов и органических катионов, тогда как почти все остальные соли хорошо растворимы в воде. Растворы солей HBF4 и таких металлов, как K, Na и т. п., имеют кислую реакцию, что указывает на их частичный гидролиз по схеме:
BF4’+ H2O Ы HF + [HOBF3]’.
При обычных условиях константа гидролиза равна 2·10-3.
Хорошо растворимы фторобораты Sn и Pb используются для электролитического рафинирования (очистки) этих металлов. Образующийся при пропускании N2O3 в концентрированную HBF4 фтороборат нитрозила NOBF4 представляет собой бесцветные твёрдые кристаллы. При нагревании с фторидами Na или K он отщепляет NOF. Был получен и фтороборат нитронила — (NO2)BF4.
Интересен продукт присоединения к KBF4 серного ангидрида — белый кристаллический KBF4·4SO3 (т. пл. 65 °С с разл.) Строение его отвечает формуле K[B(FSO3)4] с F- в качестве дважды донора(к B и к S ).
Образование аналогичных фтороборатам производных типа M[BГ4] для других галогенов не характерно. Однако соли некоторых достаточно объёмных катионов [C5H5NH+, N(CN3)4+] могут быть получены для всех галогенов, а хлориды типа M[BCl4] известны также для Cs, Rb, K и NH4. Все эти соединения гигроскопичны и бурно разлагаются водой. Как правило, они бесцветны. Исключением является оранжево-красный NOBCl4 (т. пл. 24 °С). Известны также некоторые смешанные фторохлориды типа M[BF3Cl], примером которых может служить малоустойчивый жёлтый NO[BF3Cl]. Взаимодействием ВF3 с NaH были получены солеобразные продукты состава Na[HBF3] и Na[H2BF2].
При образовании галогенидами бора комплексами с другими веществами атом B выступает в качестве акцептора. Поэтому присоединяться к молекулам ВГ3 способны только молекулы, содержащие в своём составе атом с достаточно отчётливо выраженной донорной функцией.
Хорошим примером такого комплексообразования может служить легко протекающая реакция:
H3N + BF3 = H3NBF3 + 171 кДж.
Образующаяся молекула характеризуется следующими структурными параметрами: d(NB) = 160, d(BF) = 138 пм, РNBF = 107°, РFBF = 111°. Бесцветный кристаллический H3NBF3 (т. пл. 162 °С) не растворяется в неполярных растворителях, но хорошо растворим в воде (примерно 1:3 по массе), причём лишь медленно реагирует с ней по схеме:
H3NBF3 + H2O = NH4• + [HOBF3]’.
Выше 125 °С он начинает медленно разлагаться на нитрид бора и фтороборат аммония:
4 H3NBF3 = NB + 3 NH4BF4.
В жидком аммиаке (растворимость около 1:10 по массе) образуются нестойкие продукты присоединения 1, 2 и № молекул NH3 (за счёт водородных связей по схеме NH3••• H3NBF3), а под действием амида калия протекает реакция:
H3NBF3 + 3 KNH2 = 3 KF Ї + B(NH2)3 + NH3
с образованием нестойкого амида бора. Последний сразу получается при взаимодействии с жидким аммиаком хлорида бора
BCl3 + 6 NH3 = 3 NH4Cl + B(NH2)3,
а его иодид даёт белый осадок имида бора:
2 BI3 + 9 NH3 = 6 NH4I + B2(NH)3.
Первой стадией реакции в обоих случаях является, вероятно, присоединение NH3 к молекуле BГ3. В отличие от аммиака с NСl3 (и NH2Cl) бортрифторид не взаимодействует.
Известно много различных продуктов присоединения к BF3. Некоторые из них имеют определённое значение. Так, взаимодействие ClF3 с BF3 был получен бесцветный [ClF2][BF4] (т. пл. 30 °С). Известен и [FCl2][BF4] устойчивый лишь ниже -127 °С. Охлаждение смеси BF3 + FСlO2 ведёт к образованию неустойчивых при обычных условиях бесцветных кристаллов [ClO2][BF4]. Интересен бесцветный кристаллический NH4BF4, при нагревании устойчивый до 240 °С, но чрезвычайно химически активный и полностью разлагаемый водой (с выделением кислорода). Ксенонгексафторид образует с BF3 белый, очень гигроскопичный и способный возгоняться в вакууме [XeF5][BF4] (т. пл. 90 °С). В результате взаимодействия дифтор диоксида с BF3 при низких температурах по схеме:
2 O2F2 + BF3 = 2 O2[BF4] + F2
образует фтороборат “диоксигенила” O2+. Вещество это медленно при 0 °С и быстро при обычных температурах разлагается по схеме:
2 O2[BF4] = 2 BF3 + 2 O2 + F2,
а с диоксидом азота даёт фтороборат нитронила:
2 O2[BF4] + N2O4 = 2 NO2[BF4] + 2 O2.
От оксида триметиламина производится легко гидролизующийся (CH3)3NBF3, взаимодействием которого с HF может быть получен хорошо растворимый в воде и спирте [(CH3)3NOH]BF4. Аналогичное по составу производное гидроксиламина — F2BNH2OH — имеет характер слабой одноосновной кислоты (К = 3·10-8); его калийная соль — [F3BNH2O]K — хорошо растворима в воде и спирте. Интересна способность BF3 присоединяться к некоторым комплексным цианидам. Например, известен K4[Mo(CN)8]·8BF3, который является, по-видимому, солью “двухслойного” комплексного аниона [Mo(CNBF3)3]4-. По данным инфракрасной спектроскопии, в смесях BF3 с азотом частично образуется комплекс N2® BF3.
Если наиболее типичные и многочисленные продукты присоединения бортрифторида являются фтороборатами, то у остальных галогенидов ВГ3 аддукты, как правило, образуются путём взаимодействия с бором центрального элемента донорной молекулы. Для BCl3 продуктов присоединения известно гораздо меньше, чем для BF3, для BBr3 — ещё меньше, а для BI3 — совсем мало. Примером последних может служить I3PBI3, осаждающийся при сливании сероуглеродных растворов PI3 и BI3. Этот оранжевый аддукт возгоняется в вакууме при 100 °С, тогда как тоже оранжевый Br3PBI3 устойчив до 80 °С, а желтоватый Cl3PBI3 — лишь до 35 °С. Интересно резкое различие длин связей N®B в CH3CNBCl3 (156 пм) и CH3CNBF3 (163 пм).
Продукты присоединения к галогенидам BГ3 обладают различной устойчивостью: некоторые из них, например H3PBCl3 (т. пл. 121 °С под давлением 14 атм), разлагаются лишь при нагревании, другие, например Cl3PBCl3 (т. пл. -94 °С) могут существовать только при низких температурах. Та или иная устойчивость зависит как от природы присоединяющейся молекулы [например она изменяется по рядам (CH3)3N > (CH3)2O > CH3F или (CH3)3P > (CH3)2S > CH3Cl, а также (CH3)3N > (CH3)3P > (CH3)3As > (CH3)3Sb или (CH3)2O >(CH3)2S >(CH3)2Se >(CH3)2Te], так и от природы галогена в BГ3. На нескольких различных системах (например, продуктах присоединения аминов) было показано, что по ряду F-Cl-Br-I она не уменьшается (как то считалось ранее), а возрастает.
Подобно бору, трёхвалентный азот также характеризуется координационным числом, равным четырём. Однако образуемые обоими элементами комплексы при одинаковости структурного типа имеют разный электрохимический характер: бор образует анионы [BF4]-, а азот — катионы [NH4]+. Так как у промежуточного между ними элемента — углерода — координационное число совпадает с валентностью, его соответствующие производные электронейтральны и представляют собой переходные случаи, что видно из приводимого сопоставления: Na[BF4] - [CF4] - [CH4] - [NH4]F.
С водородом бор практически не соединяется, однако при действии кислот на сплавы бора с магнием, помимо свободного водорода, выделяются небольшие количества смеси различных бороводородов (боратов), среди которых преобладает отвечающий формуле В4Н10. Последний легко распадается на В2Н6 и ряд других боранов, более бедных водородом. Простейшие бораны бесцветны и очень ядовиты. По физическим свойствам они похожи на углеводороды и силаны аналогичного состава, как это видно из приводимого ниже сопоставления точек плавления и кипения (°С):
С2Н6 | В2Н6 | Si2H6 | C4H10 | B4H10 | Si4O10 | |
| Точка плавления | -172 | -165 | -132 | -138 | -120 | -84 |
| Точка кипения | -88 | -93 | -14 | +16 | 107 |
По химическим свойствам простейшие бораны похожи на силаны. Так же как последние (и в ещё большей степени), они при обычных условиях неустойчивы. В частности, водой бараны постепенно разлагаются с выделением водорода по реакции, например:
В2Н6 + 6 Н2О = 6 Н2 + 2 Н3ВО3,
а получаемая при разложении кислотами сплавов бора с магнием газовая смесь на воздухе самовоспламеняется. Горение боранов сопровождается выделением огромного количества тепла (например, 2027 кДж/моль В2Н6 против 1425 кДж/моль С2Н6), что создаёт возможность их эффективного использования как реактивного топлива.
Для лабораторного получения небольших количеств бороводородов сплав бора с избытком магния обычно обрабатывают 8 н. раствором H3PO4. Друг от друга бораны могут быть отделены фракционной перегонкой (в отсутствии воздуха). Получение диборана B2H6 можно вести и действием электрического разряда на смесь паров BСl3 с водородом под уменьшенным давлением). Удобным методом получения диборана является проводимая в эфирной среде реакция по схеме:
6 MH + 8 BF3 = 6 MBF4 + B2H6
(где M — Li или Na). Образование диборана происходит также при пропускании смеси пара BСl3 с водородом над нагретыми металлами (Al, Mg, Zn, Na) или при взаимодействии паров галогенидов BГ3 с гидридами наиболее активных металлов (NaH, CaH2). Имеется указание и на возможность образования B2H6 около 1000 °С непосредственно из элементов.
Будучи изолирован от воздуха и воды, B2H6 может сохраняться почти без разложения месяцами. Лишь медленно идёт в этих условиях разложение и наиболее неустойчивого борана — B4H10. Продуктами его распада являются водород и другие бороводороды. Первоначально он идёт, вероятно, с отщеплением водорода и образованием более бедных им боранов, а нахождение в продуктах разложения B2H6 объясняется вторичной реакцией взаимодействия ещё не разложившегося B4H10 с водородом в момент выделения. Подобное протекание процесса косвенно подтверждается тем, что добавленный к B4H10 при его распаде Si2H6 полностью переходит в SiH4.
Обычным исходным веществом для получения остальных бороводородов является в настоящее время B2H6. Соответственно регулируя условия его термического разложения, удаётся непосредственно или через промежуточные стадии получать другие желаемые бораны. Основные направления таких переходов показаны на рис. 4.
60°С
![]()
![]()
![]()
200°С
![]()
![]()
![]() В4Н10 В5Н9
В4Н10 В5Н9
![]()


![]()
100°С 180°С

 95°С В2Н6
95°С В2Н6
180°С 120°С
![]()
![]() В10Н14 В5Н11
В10Н14 В5Н11
![]() 25°С
25°С
+Н2
![]()
100 °С
Рис. 4. Термические превращения боранов.
Помимо температуры, большое влияние на ход термических реакций боранов оказывают различные другие факторы (давление и пр.). Для использования в составе реактивных топлив наиболее перспективны В5Н9 и В10Н14. По бороводородам имеются обзорные статьи и специальная монография.
Лучше других изучены шесть бороводородов, температуры плавления и кипения которых приводятся ниже:
В2Н6 | В4Н10 | В5Н11 | В5Н9 | В6Н10 | В10Н14 | |
Температура плавления, °С | — 165 | — 120 | — 122 | — 47 | — 62 | + 99 |
Температура кипения, °С | — 93 | 18 | 63 | 60 | 108 | 213 |
Бороводороды B5H11, B5H9, и B5H10 при обычных условиях жидкие, B10H14 представляет собой летучие без разложения бесцветные кристаллы (давление пара 0,045 мм рт. ст. при 25 °С). Все эти бораны имеют отвратительный запах. Даже незначительные количества их паров в воздухе вызывают при вдыхании головную боль и тошноту.
Бороводороды являются нервными ядами. В организм они могут попадать не только через дыхательную систему, но и путём всасывания кожей. Минимально определяемое по запаху содержание их в воздухе имеет порядок тысячных долей мг/л, что уже превышает токсичную концентрацию. Острое отравление может вызвать головную боль, тошноту, слабость, судороги, состояние сильного раздражения или, наоборот, психической депрессии. При хроническом отравлении страдают главным образом органы дыхания, печень и почки. В качестве мер индивидуальной защиты рекомендуются резиновые перчатки и специальные противогазы (с гопкалитом, силикагелем и алюминием в качестве фильтрующей массы). При случайном попадании борана на кожу её следует тотчас же протереть разбавленным раствором NH4OH.
Во многих органических растворителях бораны, подобно силанам, растворяются без разложения, а водой они разрушаются быстрее силанов. Растворы щелочей разрушают бораны с выделением одной молекулы Н2 на каждую связь В-В или В-Н.
При отсутствии примесей пары перечисленных боранов (за исключением “нестабильного пентаборана” — B5H11) в сухом воздухе не самовоспламеняются. Однако во влажном воздухе такое самовоспламенение может произойти со взрывом. Вполне устойчив на воздухе при обычных температурах лишь декаборан — В10Н14. Теплота его плавления равна 33 кДж/моль, а плотность снижается при плавлении от 0,92 до 0,78 г/см3. У других боранов она в жидком состоянии колеблется от 0,45 (В2Н6) до 0,70 (В6Н10). При сопоставимых условиях индивидуальная термическая устойчивость боранов изменяется в ряду В10Н14 > В5Н9 > В2Н6 > В5Н11 > В4Н10. Термическое разложение боранов может быть использовано для борирования металлических поверхностей, что ведёт к повышению их твёрдости и химической стойкости.
По отношению к образующим их элементам бораны являются слабо эндотермичными соединениями (например, 38 кДж/моль В2Н6). Критическая температура диборана равна +17 °С, критическое давление — 40 атм. Молекула его характеризуется ионизационным потенциалом 11,4 В и неполярна. Напротив, молекулы других изученных в этом отношении боранов полярны.
Различным более или менее сложными путями были получены (частью — лишь в очень малых количествах) и некоторые другие, пока ещё мало изученные бораны: В6Н12 (т. пл. -82 °С, давление пара 17 мм рт. ст. при 0 °С и 67 при 25 °С)), В8Н12 (т. пл. 20), В9Н15 (т. пл. +3 °С, давление пара при 28 °С только 0,8 мм рт. ст.), В16Н20 (т. пл. 99 °С), В18Н22. последний известен в двух изомерных формах с температурами плавления 180 °С (норм.) и 129 °С (изо-). Имеются также отдельные указания на возникновение в определённых условиях ещё некоторых соединений того же класса. Например, среди продуктов реакций в электроразряде был обнаружен В10Н16, а из промежуточных продуктов термического разложения декаборана может быть выделен В20Н24. Сообщалось и о получении В8Н16, В8Н18, В10Н18.
Так как бор трёхвалентен, его максимально насыщенные водородом гидриды должны были бы отвечать общей формуле BnHn+2, т. е. иметь составы BH3, B2H4, B3H5, B4H6 и т. д. Однако летучие бораны такого состава неизвестны.
Молекулы летучих бороводородов следует рассматривать как результат сочетания друг с другом приведённых выше валентно-насыщенных структур при посредстве мостиковых водородных связей В···Н···В. Сочетания двух таких структур дают бораны типа BnHn+4 (в частности, B2H6, B5H9, B6H10, B10H14, B18H22), а сочетание трёх структур — бораны типа BnHn+6 (в частности, B4H10, B5H11, B6H12, B9H15, B10H16).
Состав простейших летучих бороводородов может быть “набран” только однозначно: B2H6 = BH3 + BH3 и B4H10 = BH3 + B2H4 + BH3. Однако уже для пентаборанов возможна “изомерия набора”. Так, B5H9 может строится из B3H5 + B2H4 или из B4H6 + BH3, а B5H11 — из B2H4 + B2H4 + BH3 или из BH3 + B3H5 + BH3. По мере роста n в формуле бороводорода число принципиально допустимых вариантов такого набора возрастает (например, для B10H14 их пять). Параллельно увеличивается и число принципиально возможных вариантов сочетания исходных валентно-насыщенных структур посредством водородных мостиков. Кроме того, начиная с B4H6 становится возможной изомерия самих этих исходных структур (прямая или разветвлённая цепь атомов бора), число вариантов которой быстро растёт по мере повышения n. В результате, потенциальные возможности структурной изомерии бороводородов совершенно несравнимы с нашими фактическими сведениями о ней. Обусловлено это главным образом малой устойчивостью большинства боранов.
Наиболее детально изучена молекула диборана. Она содержит два водородных мостика и может быть изображена формулой:
![]()
![]() Н
Н
![]()
![]() Н Н
Н Н
![]()
![]()
![]()
![]() В В
В В
Н Н
Н
Структура эта характеризуется расстояниями d(BB) = 178, B-H(внешн) = 120, B···H(внутр) = 133 пм и углами РНВН(внешн) = 121°, РНВН(внутр) = 96 °С. Атомы бора и крайних водородов расположены в одной плоскости, а водородные мостики — перпендикулярно к ней. Геометрически диборан представляет собой два тетраэдра из атомов водорода с общим ребром [d(HH) = 198 пм] и атомами бора, смещёнными на 15 пм от центров тетраэдров по направлению наружу. Атомы бора поляризованы положительно, ковалентно связанные водороды — слабо отрицательно, а мостиковые водороды — более отрицательно (для каждого из них предлагался эффективный заряд -0,22). Энергия обычной ковалентной связи BH(внешн) оценивается в 380 кДж/моль, а работа разрыва молекулы B2H6 на два радикала BH3 составляет 247 кДж/моль. Следовательно энергии образования каждой мостиковой связи по схеме: В-Н + В = В ···Н··· В равна 123 кДж/моль, а её
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()


![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() В Н2В Н ВН
В Н2В Н ВН
![]()

![]()
![]()
![]() Н
Н
![]() Н
Н
![]()
![]()
![]()


![]()
![]()
![]()
![]() Н Н Н
Н Н Н
![]() В
В
![]()
![]()

![]()
![]()
![]() В
В
![]()

![]() Н Н
Н Н
![]()
![]() НВ Н ВН2
НВ Н ВН2
Н
![]()
![]() В
В
Н Н
Рис. 5. Пространственная структура В4Н10. Рис. 6. Схема строения В4Н10.
полная энергия составляет 504 кДж/моль. Возникновение мостиковых связей и стабилизирует молекулы летучих бороводородов.
Строение более сложных боранов изучалось главным образом с помощью рентгеноструктурного анализа, причём в основу расшифровки полученных экспериментальных данных была положена идея заполнения атомами бора углов одной их двух геометрических фигур — октаэдра или икосаэдра. Установленная таким путём пространственная структура молекулы В4Н10 показана на рис. 5. Ей соответствует валентная схема рис. 6. Каждый атом бора имеет характерное для него координационное число 4. Длина ковалентной связи В-В равна 171 пм.
Структура молекулы В5Н9 показана на рис. 7, они говорят в пользу набора по типу В3Н5 + В2Н4 (рис. 8). Центральный атом В имеет в этом случае также характерное для бора (например, в ВГ3) координационное число 3.




![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() Н
Н
![]()
![]()
![]()
![]()


![]()
![]() В
В
![]()
![]()
![]()
![]()

![]()

![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() НВ Н ВН
НВ Н ВН

![]()
![]()
![]()
![]()
![]() Н Н
Н Н


 В
В

![]() В Н В Н
В Н В Н
![]()
![]()
![]() В В Н
В В Н
Н
Н Н Н
![]()
![]() Н НВ Н ВН
Н НВ Н ВН
Рис. 7. Пространственная структура В5Н9. Рис. 8. Схема строения В5Н9.
Неясной частью теории строения летучих боранов является и трактовка характерных для них водородных мостиков. Связь В ···Н··· В (которую ввиду её изогнутости иногда называют “банановой”) несомненно имеет иную природу, чем обычная водородная. Так как для связывание трёх атомов в ней есть лишь два электрона, она могла бы строиться по типу двух одноэлектронных связей. Такая трактовка хорошо согласуется с её относительной прочностью (которую, по примеру Н2+, для двух одноэлектронных связей можно ожидать несколько большей, чем для одной двухэлектронной). При антипараллельности спинов обоих электронов она не противоречит и диамагнетизму боранов.
Обычной является в настоящее время трактовка данной связи, как трёхцентровой с позиций теории молекулярных орбиталей: линейная комбинация трёх атомных орбиталей (по одной от каждого из атомов) даёт три молекулярные орбитали — связывающую, несвязывающую и разрыхляющую, из которых наиболее энергетически выгодная — связывающая — и заполняется единственной электронной парой. При истолковании электронодефицитных структур более сложных бороводородов также пользуются представлениями о трёх- и более центровых связях (например, пятицентровой у одного из атомов бора в В5Н9).
Отличный от других бороводородов характер имеет первоначально синтезированный в электроразряде В20Н16: в его структуре нет мостиковых связей. Он представляет собой бесцветное кристаллическое вещество (т. пл. 199 °С), способное уже выше 100 °С возгоняться в вакууме, гигроскопичное и реагирующее с водой, образуя сильную двухосновную кислоту. Вместе с тем известны (но ближе не изучены) ещё более бедные водородом твёрдые бораны, различающиеся своей окраской (в большинстве случаев жёлтых оттенков) и растворимостью в сероуглерода. Коричневый цвет наиболее бедных водородом боранов приближается к цвету аморфного бора.
Для трактовки химических свойств летучих бороводородов основное значение имеет то обстоятельство, что атомы бора не является полностью экранированными. В результате присоединения к ним тех или иных частиц из окружающей Среды вероятны, вообще говоря, два основных направления первичного элементарного процесса: разрыв водородных мостиков или миграция протонов, приводящая к отщеплению Н2 и образованию ковалентных связей В-В. Выбор того или иного направления зависит от свойств присоединяющихся частиц. Так как полнее изучены химические превращения диборана, они преимущественно и рассматриваются ниже.
При действии ультрафиолетовых лучей (с длиной волны 184,9 нм) диборан параллельно испытывает два типа распада — В2Н6 = B2H5 + Н и В2Н6 = ВН3 + ВН3 — причём вероятности осуществления того или иного относятся приблизительно как 10:1. В смеси диборана с бортрихлоридом при обычных температурах устанавливается смещённое влево равновесие по схеме:
В2Н6 + ВCl3 = В2Н5Cl + ВНCl2
Первичной стадией взаимодействия диборана с водой является разрыв мостиковых связей:
Н2О + Н2В(НН)ВН2 + ОН2 = 2 Н2ОВН3.
Вслед за тем протон из молекулы воды мигрирует в отрицательно поляризованный водород связи В-Н, что сопровождается отщеплением молекулы водорода:
Н2ОВН3® Н2 + НОВН2.
Далее тот же процесс дважды повторяется по схеме:
Н2О + ВН2(ОН) Ы Н2ОВН2(ОН) Ы Н2 + НВ(ОН)2Ы Н2 + (НО)3В и
Н2О + ВН(ОН)2Ы Н2 + (НО)3В.
Так как у каждого атома бора в промежуточных продуктах гидролиза — НОВН2 и (НО)2ВН — занято лишь по три координационных места, вероятность присоединения к ним молекул воды больше, чем к атомам бора в диборане. Отсюда и бульшая скорость рассматриваемых вторичных реакций по сравнению с первичной, из-за чего промежуточные продукты гидролиза не накапливаются. При -130 °С для диборана был получен кристаллогидрат состава В2Н6·2Н2О с вероятной структурой 2Н2ОВН3, что соответствует первой стадии приведённого выше хода гидролиза.
Гидролитическое разложение остальных бороводородов идёт гораздо медленнее, чем диборана. До известной степени это обусловлено их малой растворимостью в воде. Так, было показано, что взаимодействие В5Н9 и Н2О по суммарной схеме:
В5Н9 + 15 Н2О = 12 Н2 + 5 В(ОН)3
несравненно быстрее протекает в диоксановом растворе обоих веществ, чем при их непосредственном контакте.
Подобно силанам, бораны не взаимодействуют с концентрированной серной кислотой. С сильными щелочами идёт реакция, не имеющая аналогии в химии кремния: помещённая в атмосферу В2Н6 палочка КОН покрывается белой коркой кристаллического гипобората калия, образующегося по схеме:
КОН + Н2В(НН)ВН2 + КОН = 2 К[HOBH3].
Твёрдые гипобораты натрия и калия устойчивы. На воздухе они притягивают влагу и расплываются, причём начинает протекать их последовательный гидролиз по схемам:
К[ВН3OН] + H2О = К[(HO)2BH2] + H2, затем
K[ВН2(OH)2] + Н2O = К[BH(OH)3] + H2
и наконец K[ВН(OH)3] + Н2O = К[B(OH)4] + H2.
В присутствии большого избытка щёлочи самопроизвольное разложение гипоборатов протекает очень медленно, но при разбавлении раствора водой оно значительно ускоряется. Ещё быстрее, практически моментально, идёт распад при подкислении раствора. Гипобораты и продукты их частичного гидролиза являются исключительно сильными восстановителями. Так, из солей Ag, Hg, Bi и Sb они выделяют свободные металлы.
Взаимодействие с растворами щелочей жидких боранов идёт несколько иначе, чем В2Н6. Первоначально они растворяются без выделения водорода. При подкислении раствора начинается распад на Н2 и Н3ВО3, однако он идёт весьма медленно и реакция для своего завершения требует многих дней. В щелочных растворах жидких боранов образуются соли более устойчивых кислот.
Возможно, что они тождественны солям, получаемым путём обработки борида магния щелочами. Соли эти (а также одновременно получаемые производные Н2В4О6) представляют собой бесцветные кристаллические вещества, растворы которых являются сильными восстановителями.
Твёрдый при обычных условиях В10Н14 растворяется в спиртах и эфирах, причём проявляет свойства сильной одноосновной кислоты. Некоторые соли декаборана известны и в твёрдом состоянии. Так, взаимодействием NaH с раствором В10Н14 в пропиловом эфире был получен NaВ10Н13, а взаимодействием раствора В10Н14 в диглиме с водным раствором [N(CH3)4]OH — нерастворимый в воде жёлтый [N(CH3)4]В10Н13. Свойства двухосновной кислоты характерны и для обеих форм В18Н16. Примером соли последней из них может служить довольно устойчивый к гидролизу К2В10Н14 (получаемый действием КВН4 на декаборан в водной среде). Были получены также соли типов МIB11H14 M2IB11H13 (вторые — в сильнощелочной среде), производящиеся от неизвестного в свободном состоянии борана В11Н15.
Исходя из В10Н14 были получены интересные производные ионов В10Н102-и В12Н122-. Известны не только содержащие их соли (в частности, нерастворимые серебряные), но и кристаллогидраты свободных кислот — (Н3О)2В10Н10·хН2О и (Н3О)2В12Н12·хН2О. Кислоты эти сравнимы по силе с Н2SO4.
Рассматриваемые ионы могли бы производиться от нормальных насыщенных боранов В10Н12 и В12Н14, но с этим трудно совместить принятое для них при рентгеноструктурном анализе икосаэдрическое взаимное расположение атомов бора. Оба иона отличаются высокой химической стойкостью — на них практически не действуют ни кислоты, ни щелочи. Их атомы водорода могут быть частично или полностью заменены на атомы галогенов, причём химические свойства при такой замене существенно не изменяются. В частности, Cs2В10Cl10 устойчив на воздухе до 400 °С.
Из производных диборана (В2Н6) наиболее важны аналогичные фтороборатам по строению соли типа М[BH4] (боргидриды, или боранаты), известные для ряда металлов. Примером может служить бесцветный кристаллический NaBH4, устойчивый при обычных условиях и хорошо растворимый в воде. Водород в анионе {BH4]- отрицателен и играет роль атома галогена. Все боронаты являются сильными восстановителями.
Взаимодействие диборана со взвешенным в кипящем эфире порошком гидрида лития ведёт к образованию бороната лития по схеме:
2 LiH + В2Н6 = 2 Li[ВН4]
Более практически важный боранат натрия в аналогичных условиях не образуется. Для его получения предложены различные пути, например основанный на реакциях:
NaH + B(OCH3)3 = Na[HB(OCH3)3] и затем
Na[HB(OCH3)3] + B2H6 = 2 Na[BH4] + 2 B(OCH3)3.
Первая из этих последовательных реакций проводится с кипящим B(OCH3)3, а вторая легко идёт при комнатной температуре. Из Na[BH4], например, по схеме:
NaBH4 + KOH = KBH4 + NaOH
могут быть получены боранаты и других щелочных металлов. Взаимодействие NaBH4 c NH4F в жидком аммиаке был получен и устойчивый лишь ниже -40 °С боранат аммония — NH4BH4.
Боргидриды щелочных металлов имеют характер типичных солей. Ион BH4- представляет собой тетраэдр из атомов водорода с атомом бора в центре и эффективным радиусом 203 пм. Связь В-Н имеет расстояние 126 пм. Энергия образования иона BH4- из BH3 и H- оценивается в 314 кДж/моль.
В атмосфере водорода (или азота) LiBH4 плавится при 278 °С (с разл.), NaBH4 устойчив до 400 °С, а KBH4 — даже до 500 °С. Боргидриды щелочных металлов хорошо растворимы в воде (например 55 г NaBH4 на 100 г Н2О при 20 °С) и взаимодействует с ней по суммарной схеме:
MBH4 + 4 H2O = 4 H2 + MOH + H3BO3.
Однако образующаяся щёлочь тормозит протекание процесса, и поэтому он идёт не до конца. Скорость гидролиза уменьшается в ряду Li » Na > K, причём боргидриды Na и K разлагаются холодной водой лишь очень медленно, а из содержащий небольшой избыток щёлочи раствора могут быть выделены кристаллогидраты NaBH4·2H2O и KBH4·nH2O (где n = 3 или 1). Горячая вода разлагает рассматриваемые боргидриды быстрее, а в кислой среде разложение их идёт очень быстро. Параллельно с гидролизом под действием кислот может частично протекать и реакция типа:
2 MBH4 + 2 HСl = 2 MСl + H2 + B2H6.
В отличие от своих аналогов LiBH4 несколько растворим в эфире (1,3 при 0 °С и 4,5 масс. % при 25 °С). Хорошими растворителями NaBH4 являются жидкий аммиак (1:1 по массе) и “диглим” — O(CH2CH2OCH3)2 (т. кип. 161 °С). При поджигании на воздухе NaBH4 спокойно сгорает.
Боргидриды щелочных металлов обладают сильно выраженными восстановительными свойствами. Практически чаще всего используется NaBH4. Например, с его помощью удобно получать летучие гидриды Ge, Sn, As и Sb, исходя из их хлоридов. Широкое использование находит NaBH4 и в органической химии (главным образом как восстановитель альдегидов и кетонов до соответствующих спиртов). Взаимодействие его с BF3 (в эфирной среде) или с BСl3 (в диглиме) может служить удобным методом получения больших количеств диборана, а подкисленного раствора — получения водорода (до 2,37 л/г NaBH4). Ещё больше водорода (до 4,13 л/г) можно получить аналогичным путём из LiBH4. Действием на последний HCN в эфирной среде был синтезирован довольно устойчивый Li[BH3CN]. Известны и другие соли аналогичного типа, например K[BH3F], K[BH3NCS], K[BH3GeH3], Na[BH3Re(CO)5].
Боргидриды M[BH4]2 (где M — Mg, Ca, Sr, Ba) представляют собой белые твёрдые вещества. Они похожи по свойствам на боргидриды щелочных металлов, но несколько менее устойчивы по отношению к нагреванию и более активны как восстановители. Гораздо менее устойчивы и более реакционноспособны Be(BH4)2 (т. возг. 91 °С) и особенно жидкий при обычных условиях Al(BH4)3 (т. пл. -65 °С, т. кип. +45 °С). Оба эти боргидрида бурно реагируют с водой. Молекулы их построены на основе двойных мостиковых связей (по типу B2H6) между радикалами BH3 и гидридами BeH2 или AlH3 [с параметрами d(BH) = 128, d(BeH) = 163, d(BeB) = 174, d(AlH) = 210, d(AlB) = 215 пм].
Были описаны и боргидриды некоторых других элементов. Из них к числу относительно устойчивых могут быть отнесены белые Zr(BH4)4 (т. пл. 29, т. кип. 128 °С) и Hf(BH4)4 (т. пл. 29, т. кип. 118 °С), а к числу наиболее неустойчивых — разлагающихся уже выше -65 °С жёлтыйSn(BH4)2, выше -30 °С бесцветныйAgBH4, выше 0 °С бесцветный Cd(BH4)2, выше 25 °С зелёный Ti(BH4)3 и выше 85 °С бесцветный Zn(BH4)2. Интересным боранатом является устойчивый лишь при низких температурах жёлтый [Mn(CO)5]BH4 (т. пл. -78 °С).
При взаимодействии B2H6 с амальгамой натрия в эфирной среде реакция идёт по уравнению:
2 Na + 2 B2H6 = NaBH4Ї + NaB3H8.
Путём испарения эфира последняя соль может быть выделена. Помимо растворимости в эфире, она отличается от NaBH4 и несколько большей устойчивостью по отношению к воде. Получены были и некоторые другие соли того же аниона (в частности, TlB3H8). Структура иона B3H8- отвечает сочетанию иона BH4- с молекулой B2H4 посредством двух водородных связей.
Растворённый в диглиме NaBH4 способен взаимодействовать с избытком диборана по схеме:
2 NaBH4 + B2H6 = 2 NaB2H7.
Для иона B2H7-была предложена структура [H3B ···H··· BH3] с одной мостиковой связью между группами BH3 (энергия которой оценивается в 125 кДж/моль). Такой трактовке формально отвечает комплексный ион HF2-.
По отношению к свободному хлору и брому бороводороды ведут себя аналогично силанам (и углеводородам): оба галогена не присоединяются в боранам, а замещают в них атомы водорода. При избытке диборана реакция идёт по схеме:
В2Н6 + Г2 = В2Н5Г + НГ,
причём могут образовываться продукты замещения на галогенид и более чем одного атома водорода. При избытке галогена происходит полное замещение:
В2Н6 + 6 Г2 = 2 ВГ3 + 6 НГ.
Подобно кремневодородам в присутствии соответствующих галогенидов Al бораны обменивают водород на галоген и при взаимодействии со свободными галогенводородами, например, по схеме:
В2Н6 + НГ = В2Н5Г + Н2.
Иодистый водород реагирует подобным же образом и без катализатора, что обусловлено наибольшей лёгкостью отщепления протона именно им.
Продукт этой реакции — B2H5I представляет собой бесцветную жидкость, затвердевающую при -110 °С. Под действием амальгамы натрия он отщепляет иод с образованием B4H10 по схеме:
2 B2H5I + 2 Na = B4H10 + 2 NaI.
Реакция эта аналогична взаимодействию с металлическим натрием галогензамещённых углеводородов.
Монобромдиборан — B2H5Br — представляет собой бесцветный газ (т. пл. -104, т. кип. +10 °С), склонный к самопроизвольному распаду на B2H6 и BВr3 по суммарному уравнению:
6 B2H5Br = 2 BВr3 + 5 B2H6.
Ещё быстрее подобным же образом распадается соответствующий хлорид (т. пл. -143, т. кип. -11 °С) и продукты, содержащие в молекуле более одного атома галогена.
С аммиаком бороводороды дают белые солеобразные продукты присоединения. Взаимодействие диборана идёт по схеме:
2 NH3 + B2H6 = [H3NBH2NH3]BH4
(т. пл. 90 °С). Возможно, что первичным продуктом при этом является легко димеризующийся H3NBH3, который может быть получен в эфирной среде по реакции:
2 LiBH4 + (NH4)2SO4 = Li2SO4 + 2 H2 + 2 H3NBH3.
Для мономера было найдено d(NB) = 156 пм. Взаимодействие B2H6 с триметиламином ведёт к образованию устойчивого мономера (CH3)3NBH3 (т. кип. 171 °С).
При низких температурах диборан реагирует с фосфином, образуя бесцветные кристаллы H3PBH3, на воздухе самовоспламеняющиеся. Значительно устойчивее (CH3)3PBH3 (т. пл. 103 °С).
В смеси диборана с монооксидом углерода устанавливается равновесие по схеме:
OC + B2H6 + CO = 2 OCBH3 + 38 кДж/моль
Образующийся карбонилборгидрид (т. пл. -137 °С, т. кип. -64 °С) представляет собой бесцветный газ.
Нагревание H3NBH3 в запаянной трубке до 270 °С ведёт к образованию триборинтриимина — В3N3H6. Последний представляет собой бесцветное жидкое вещество (т. пл. -56, т. кип. +55 °С). Молекула В3N3H6 имеет форму шестиугольника из поочерёдно расположенных радикалов ВН и NH. Интересно, что симметричная, казалось бы, молекула В3N3H6 имеет дипольный момент. Обусловлено это отклонением связей N-H от плоскости шестиугольника (или не плоской структурой последнего). Так как триборинтриимин (иначе, боразол или боразин) по строению и некоторым физическим свойствам похож на бензол, его иногда называют “неорганическим бензолом”.
Следует отметить, что аналогия эта часто переоценивается. Так, в отличие от бензола боразол неустойчив по отношению к воздуху и медленно разлагается при хранении даже в его отсутствие. Также в отличие от бензола для В3N3H6 характерны реакции присоединения (обычно — трёх других молекул на молекулу боразола). Так присоединяются, например, HСl, HВr, H2O, CH3OH, C2H5OH. Боразол растворим в воде и гидролизуется ею (очень медленно на холоду и быстрее при нагревании) с образованием Н2, В(ОН)3 и NH4OH. В общем, разрыв его цикла осуществляется несравненно легче, чем у бензола. Например, такой разрыв легко происходит под действием щелочей.
Галогениды замещаются в боразоле только водород стоящий при атомах бора, т. е. действием хлора можно получить лишь В3N3H3Cl3. Полный хлорный аналог боразола — В3N3Сl6 (т. пл. 177 °С) удалось синтезироваться по схеме:
3 BCl3 + 3 NCl3 = 6 Cl2 + В3N3Cl6.
Реакция эта интересна как пример синтетического использования разноимённой зарядности хлора (d- в BСl3 и d + в NCl3).
Химия бороводородов и их производных по своему характеру и богатству синтетических возможностей приближается к органической химии. Она очень интересна, но трудна экспериментально и во многом необычна, так как не укладывается в рамках установившейся валентно-структурной теории, которая позволяет охватить единой трактовкой химию не только всех остальных элементов, но и неводных соединений самого бора.
Вряд ли это означает общее несовершенство наших современных теоретических представлений и необходимость их радикального изменения. Скорее можно думать, что причиной является недоучет каких-то особых обстоятельств, присущих электронодефицитных молекул.
Положение аналогично сложившемуся в конце прошлого века, когда возник кризис классической теории валентности, вызванной накоплением экспериментальных данных по “соединениям высшего порядка” (комплексным). Как известно, кризис этот был ликвидирован не путём отказа от теории валентности, а путём её дополнения координационной теорией (первоначально чисто формальной, так как физическая природа “побочных валентностей” была не ясна).
В данном случае классическая теория двухцентровых валентных связей дополняется представлением о возможности существования трёх- и более-центровых связей. Применяется это представление только к тем частям структур электронодефицитных молекул (например, мостиковым связям В ···Н··· В), которые не укладываются в рамки обычной теории валентности. Так как понятие многоцентровости является по сути дела эклектическим. Отражает ли он физическую суть дела (или представляет собой форму описания), пока не ясно.
Следует отметить, что полный переход на метод молекулярных орбиталей вопроса не решает. Прежде всего метод МО ограничен по своим практическим возможностям (так как с увеличением атомности молекул построение системы их орбиталей быстро усложняется). Вместе с тем он даёт трактовку молекулы в целом, тогда как для химии наиболее важны характеристики её отдельных атомных сочетаний (валентных связей).
Ниже сопоставлены средние длины и энергии связей бора:
| Связь | В-В | В-Н | В-О | В-S | B-N | B-C | B-F | B-Cl | B-Br | B-I |
| Длина, пм | 172 | 120 | 136 | 181 | 142 | 156 | 130 | 173 | 187 | 210 |
| Энергия, кДж/моль | 330 | 380 | 514 | 443 | 372 | 644 | 443 | 376 | 284 |
Приведённые данные относятся к простым ковалентным связям при координационном числе 3 для бора. С переходом к координационному числу 4 длины возрастают, а энергии уменьшаются. Например, длина связи В-О становится равной 147 пм, а её энергия — 401 кДж/моль. Подобным же образом средняя длина донорно-акцепторной связи N®B (образование которой вызывает повышение координационного числа бора от 3 до 4) равна 158 пм. Следует также отметить, что величина 443 кДж/моль для связи B-N относится к боразолу и его производным.
4. Подгруппа брома.
Содержание в земной коре брома составляет 3·10-5 %, а иода 4 ·10-6 %. По характеру распределения в природе оба элемента очень похожи на хлор, но образование вторичных скоплений для них не характерно. Содержание в природе астата ничтожно мало и свойства этого элемента почти не изучены.
Природный бром состоит из смеси изотопов 79Вr (50,5 %) и 81Br (49,5 %), тогда как иод является “чистым” элементом — состоит из атомов 127I. Для астата известны только радиоактивные изотопы с небольшой продолжительностью жизни атомов (в среднем 12 ч для наиболее долгоживущего 210At).
Основными источниками промышленного получения брома являются воды некоторых соляных озер (0,01-0,5 % Вr) и морская вода (в среднем 0,007 % Вr). Частично он добывается также из бромистых соединений, примеси которых обычно содержатся в природных месторождениях калийных солей, и из буровых вод нефтеносных районов (0,01-0,1 % Br).
Для промышленной добычи иода основное значение имеют именно буровые воды, содержащие в среднем 0,003%. Другим источником этого элемента является зола морских водорослей.
Для получения свободных брома и иода можно воспользоваться вытеснением их хлором. Бром выделяется из раствора исходной соли в виде тяжелой жидкости, иод — в твердом состоянии.
Иод был открыт в 1811 г., бром — в 1826 г. Существование астата предсказывалось уже Д. И. Менделеевым. Элемент этот был получен искусственно в 1940 г. Происхождение брома и иода земной поверхности такое же как хлора и фтора — основные массы обоих элементов выделялись из горячих недр Земли в форме своих водородных соединений.
При получении брома из морской (или озёрной) воды её подкисляют серной кислотой до рН = 3,5 и обрабатывают хлором. Выделяющийся бром перегоняют током воздуха в раствор соды, который после достаточного насыщения бромом подкисляют. Реакции протекают по уравнениям:
2 NаВr + Сl2 = 2 NаСl + Вr2, затем
3 Вr2 + 3 Nа2СО3 = 5 NаВr + NаВrО3 + 3 СО2 и, наконец,
5 NаВr + NаВrO3 + 3 Н2SO4 = 3 Na2SO4 + 3 Вr2 + 3 Н2О.
Технический бром часто содержит примесь хлора. Для очистки его обрабатывают концентрированным раствором СаВr2, причем хлор вытесняет бром, который при разбавлении раствора выделяется в виде тяжелого слоя, содержащего лишь очень немного (порядка 0,05 %) растворенной воды.
В безводном состоянии бром может быть получен отгонкой из смеси с концентрированной Н2SO4. Тройной точке на его диаграмме состояния отвечает температура -7,3 °С и давление 46 мм рт. ст. Жидкий бром имеет весьма низкое значение диэлектрической проницаемости (e = 3). Охлаждение его насыщенного водного раствора ведет к образованию кристаллогидрата Вr2·8Н2О (т. пл. 6 °С). Известен также нестойкий кристаллосольват с бензолом состава Вr2·С6Н6 (т. пл. -14 °С).
Так как содержание иода в буровых водах очень мало, основной задачей при получении является его концентрирование. Это обычно достигается выделением иода в свободном состоянии, чаще всего — по реакции:
2 NаI + 2 NаNО2 + 2 Н2SO4 = 2 Na2SO4 + I2 + 2 NО + 2 Н2О
с последующей его адсорбцией на активированном угле. Из последнего иод извлекают горячим раствором едкого натра по реакции:
3 I2 + 6 NаOH = 5 NаI + NаIO3 + 3 Н2О
После насыщения раствора подкислением его вновь выделяют свободный иод по реакции
5 NаI + NаIO3 + 3 Н2SO4 = 3 Nа2SO4 + 3 I2 + 3 Н2О
Морская вода содержит около 5·10-6 % иода, который извлекается из нее некоторыми водорослями и накапливается ими. Например, широко используемая населением Китая и Японии в качестве пищевого продукта ламинария (морская капуста) содержит в воздушно-сухом состоянии около 0,5 % иода.
Для получения иода из золы морских водорослей её обрабатывают водой и после упаривания раствора оставляют его кристаллизоваться. Бульшая часть содержащихся в золе хлористых и сернокислых солей выпадает при этом в осадок, а иодистые соли, как более растворимые, остаются в растворе. Иод извлекают затем обработкой раствора хлором (или МnО2 и Н2SO4).
По основным физическим свойствам бром и иод закономерно укладываются в один ряд с хлором и фтором, как это видно из приводимой ниже таблицы (в которую включен также водород):
| При обычных условиях | |||||
| Химическая формула | Молекулярный вес округленно | Агрегатное состояние | Цвет | Тпл°С | Ткип°С |
H2 | 2 | Газ | Бесцветный | -259 | -253 |
F2 | 38 | Газ | Почти Бесцветный | -220 | -188 |
Cl2 | 71 | Газ | Желто-зеленый | -101 | -34 |
Br2 | 160 | Жидкость | Темно-коричневый | -7 | 59 |
I2 | 254 | Твердое вещество | Темно-серый | 114 | 186 |
Плотность брома равна 3,1, иода 4,9 г/см3. Так как давление пара твердого иода очень велико, он при нагревании легко возгоняется. Возгонкой технического иода пользуются для его очистки.
Для температур плавления и кипения астата даются значения 227 и 317 °С. Теплоты плавления брома, иода и астата равны соответственно 10,5, 15,9 и 20,9 кДж/моль, а теплоты их испарения (при температурах кипения) — 29,7, 41,8 и 54,3 кДж/моль. Критическая температура брома равна 311, иода — 553 °С. Интересно, что давление паров брома и иода в присутствии индифферентных газов (N2 и др.) выше, чем при той же температуре без них.
Тройной точке на диаграмме состояния иода соответствует температура 116 °С и давление 90 мм рт. ст. Для получения жидкого иода необходимо, следовательно, создать такие условия, чтобы парциальное давление его паров превышало 90 мм рт. ст. Это проще всего достигается нагреванием достигается нагреванием большого количества кристаллов иода в колбе с узким горлом.
Жидкий иод имеет довольно высокое значение диэлектрической проницаемости (e = 11). Он растворяет S, Sе, Те, иодиды ряда металлов и многих органические соединения. Раствор в нем иодистого калия проводит электрический ток. Сам иод диссоциирован по схеме I2Ы I- + 1+, но диссоциация эта очень мала: [I+][I-] = 10-42.
Темно-фиолетовые пары иода и красно-коричневые пары брома (в еще большей степени) обладают резким запахом. По действию на организмы бром близок к хлору. Бром применяется главным образом для выработки специальных добавок к моторным бензинам. Иод в виде 5 %-ного спиртового раствора (“иодной настойки”) применяется для стерилизации ран. Соединения обоих тяжелых галогенов имеют большое значение для фотографии, медицины и т. д. Ежегодная мировая выработка брома исчисляется десятками тысяч тонн, иода — тысячами тонн.
Физиологическая роль бромистых соединений в нормальной жизнедеятельности организма еще недостаточно выяснена. К их дополнительному введению наиболее чувствительна центральная нервная система: бромиды используются в медицине как успокаивающие средства при повышенной возбудимости. Чрезмерное их накопление способствует появлению кожных сыпей. Выводятся они из организма очень медленно (главным образом, с мочой). По токсическому действию паров бром похож на хлор. При ожоге кожи жидким бромом рекомендуется промыть пострадавшее место разбавленным раствором аммиака.
Соединения иода играют важную роль в регулировании обмена веществ. У животных организмов иод накапливается главным образом в щитовидной железе (аналогично ведет себя и вводимый в организм астат). Тело человека содержит около 25 мг иода, из которых примерно 15 мг находится в щитовидной железе. Из обычных продуктов питания наиболее богаты иодом лук и морская рыба. Недостаток иода служит причиной болезни, известной под названием “зоба”. Болезнью этой иногда страдает поголовно все население тех местностей (главным образом удаленных от моря возвышенностей), в которых воздух, вода и пища содержат слишком мало иода. Ежедневное потребление небольших — порядка 0,1 мг — доз иодидов (в виде примеси к поваренной соли) позволяет полностью избавиться от этой болезни. В Китае больных зобом издавна лечили золой морских губок (которая содержит до 8,5% иода). При добавлении в пищу иодсодержащих водорослей у коров увеличивается удой молока, а у овец быстрее растет шерсть. Отмечено также благотворное влияние небольших доз иодистых соединений на яйценоскость кур, откорм свиней и т. д.
Широко применяемая “иодная настойка” может быть приготовлена смешиванием в равных долях 10 %-ного раствора иода в спирте (95 %) и 4 %-ного водного раствора KI. Добавка иодистого калия повышает устойчивость жидкости при хранении. Следует отметить, что не только сам иод, но и многие его соединения (в частности, KI) хорошо всасываются организмом даже через неповреждённую кожу. Приём иодной настойки внутрь (1-5 капель на молоке) назначается иногда при атеросклерозе. Избыточное поступление иода в организм может вызвать некоторые неприятные явления (насморк, кожные сыпи и т. д.), исчезающие при прекращении приема иода.
Растворимость брома в воде составляет около 35 г, а иода — 0,3 г на литр. Оба эти галогена (и астат) гораздо лучше растворяются в различных органических растворителях.
Растворимость иода в воде сильно возрастает с повышением температуры и при 100 °С достигает 3,3 г/л. Органические жидкости растворяют его значительно лучше воды, как то видно из приводимых ниже примерных данных (в вес.% при обычных условиях):
С2H5OH | (C2H5)2O | C6H6 | CHCl3 | CCl4 | CS2 | ||
| 20 | 24 | 12 | 2,5 | 2,5 | 13 |
Растворы иода в разных растворителях имеют различные окраски: фиолетовую, красную, коричневую и промежуточных оттенков. Так как состоящие из свободных молекул I2 пары иода характеризуются сами по себе синей, а в смеси с воздухом фиолетовой окраской, наличие последней в растворе (например, в ССl4 или НГ) указывает на отсутствие заметной сольватации растворенных молекул иода. Напротив, коричневый цвет раствора (например, водного или спиртового) указывает на сильную сольватацию. В отличие от иода, цвет растворов брома почти не зависит от природы растворителя.
Благодаря лучшей, чем в воде, растворимости галоидов в органических растворителях, при соприкосновении водного раствора с органическим растворителем бульшая часть галогена переходит в последний. При этом галоген распределяется между органическим растворителем и водой в строго определенных отношениях. Если в качестве примера взять бром и сероуглерод (СS2), то отношение концентрации брома в сероуглеродной фазе к концентрации его в водной при различных общих количествах растворенного брома остается постоянным и равным примерно 80.
В этом постоянстве отношения концентраций (точнее, отношения активностей) распределение между двумя несмешивающимися растворителями вещества заключается так называемый закон распределения. Он верен, однако, лишь в том случае, если распределяемое вещество в обеих фазах имеет один и тот же состав (например из молекул) и не вступает в прямое химическое взаимодействие с растворителем. Найденное отношение концентраций (в данном примере 80) называется коэффициентом распределения. Величина его (при постоянной температуре) характерна для данной системы: растворитель А — распределяемое вещество — растворитель Б. Например, при замене сероуглерода на ССl4 коэффициент распределения брома становится равным примерно 30. Распределение имеет большое техническое значение, так как часто позволяет избирательно извлекать (экстрагировать) то или иное вещество из раствора смеси веществ.
По своей наиболее характерной химической функции бром и иод являются одновалентными неметаллами. Некоторые числовые характеристики обоих элементов сопоставлены ниже с аналогичными данными для хлора и фтора (Г — общее обозначение галогена):
Молекула Г2 | Ядерное расстояние пм | Энергия Диссоциации кДж/моль | Атом Г | Эффективный радиус, пм | Сродство к электрону, кДж/моль | Ион Г | Эффективный радиус, пм | Энергия гидратации, кДж/моль |
F2 | 142 | 159 | F | 71 | 339 | F- | 133 | 485 |
Cl2 | 198 | 242 | Cl | 99 | 355 | Cl- | 181 | 351 |
Br2 | 229 | 192 | Br | 114 | 330 | Br- | 196 | 318 |
I2 | 267 | 150 | I | 133 | 301 | I- | 220 | 280 |
Химическая активность брома и иода меньше, чем у хлора, но все же велика. Со многими металлами и некоторыми неметаллами (например, фосфором) они способны взаимодействовать в обычных условиях. При этом бром по активности мало уступает хлору, тогда как иод отличается от него уже значительно.
Подобно атомам фтора и хлора, в основном состоянии атомы брома (4s24р5) и иода (5s25р5) одновалентны.
При выводе количественных характеристик сравнительной металлоидной активности галоидов в отсутствие воды вместо энергий гидратации должны учитываться энергии связей (в ковалентных системах) или энергии кристаллических решеток (в ионных системах). Как показывает приводимое ниже примерное сопоставление, все эти величины изменяются приблизительно однотипно:
| F | Cl | Br | I | |
Энергии гидратации ионов Г-, кДж/моль | 485 | 351 | 318 | 280 |
Энергии связей С-Г, кДж/моль | 485 | 339 | 284 | 231 |
| Энергии решеток NaГ, кДж/моль | 915 | 777 | 740 | 690 |
Поэтому общий характер изменения металлоидной активности по ряду F-С1-Вr-I остается неизменным.
На образовании и последующем термическом разложении летучих иодидов основано иодидное рафинирование некоторых металлов (Сr, V, Тi и др.) Проводится оно в замкнутой системе путем взаимодействия иода с технически чистым образцом при 100-500 °С под давлением порядка 10-4 мм рт. ст., причем пары образующегося иодида тут же термически разлагаются на поверхности нагретой до 1300-1500 °С проволоки. Иод вновь вступает в реакцию, а вокруг проволоки постепенно наращивается стержень обрабатываемого металла, свободного от нелетучих при условиях опыта примесей.
Взаимодействие брома с водородом происходит лишь при нагревании. Иод с водородом реагирует только при достаточно сильном нагревании и не полностью, так как начитает идти обратная реакция — разложение иодистого водорода. Оба галогеноводорода удобно получать разложением водой соответствующих галогенидных соединений фосфора по схеме:
РГ3 + 3 Н2О = Н3РО3 + 3 НГ
Реакция легко идет уже при обычной температуре.
Синтез НВr из элементов протекает при 200-300 °С с измеримой скоростью по следующим уравнениям:
Вr2 + 192 кДж = 2 Вr (первоначальное возбуждение),
Вr + Н2 = НBr + Н,
затем Н+ Вr2 = НBr + Вr и т. д.
В отличие от синтеза НСl вторая реакция затруднена из-за её эндотермичности (71 кДж/моль), а обратная ей реакция
Н + НВг = Н2 + Вr
протекает легко. Поэтому возникающие цепи часто обрываются и процесс не приобретает взрывного характера. Так как реакция I + Н2 = НI + Н ещё более эндотермична (138 кДж/моль), синтез HI вообще не является цепной реакцией, а протекает по обычному бимолекулярному типу.
Подобно хлористому водороду, HBr и HI представляют собой бесцветные газы, очень хорошо растворимые в воде. Некоторые их свойства сопоставлены со свойствами HF и HCl в приводимой ниже таблице. По ряду НI-НВr-НСl свойства изменяются весьма закономерно, тогда как при дальнейшем переходе к НF наблюдается более или менее резкий их скачок, иногда даже в направлении, обратном общему ходу. Обусловлено это сильной ассоциацией фтористого водорода, отсутствующей у его аналогов.
Энергии связей Н-Вr и Н-I равны соответственно 364 и 297 кДж/моль. Жидкие галоленоводороды характеризуются при температурах кипения плотностями 2,2 (НВr) и 2,8 (НI) г/см3 и теплотами испарения 17,6 и 19,6 кДж/моль. Как растворители, они похожи на НСl. Энергии диссоциации молекул НГ на свободные газообразные ионы Н+ и Г- составляют 1517 (НF), 1359 (НСl), 1317 (НВr) и 1283 (НI) кДж/моль. Теплота образования АtН из элементов оценивается в –105 кДж/моль.
Судя по характеру изменения теплот образования гидрогалогенидов, их термическая устойчивость должна сильно уменьшаться от фтора к иоду. Действительно, распад НF на элементы становится заметен лишь выше 3500 °С, тогда как для других галоидоводородов имеем при 1000 °С следующие степени диссоциации: 0,0014 (НС1), 0,5 (НВг) и 33 % (НI). В органических растворителях (бензоле и т. п.) все гидрогалиды растворимы гораздо хуже, чем в воде.
Как и хлористый водород, НВr и НI образуют с водой азеотропные смеси, содержащие соответственно 47 % НВr (т. кип. 126 °С) и 57 % НI (т. кип. 127 °С). Для обоих галогеноводородов известны кристаллогидраты с 2, 3 и 4 молекулами воды. И для брома, и для иода были получены аналогичные соответствующему хлориду нестойкие производные типа [ХR4]НГ2, где R — органический радикал.
Увеличение электролитической диссоциации при переходе от НF к НI обусловлено, вероятно, уменьшением поверхностной плотности отрицательного заряда галоидов в связи с ростом их ионных радиусов.
В неводных растворителях галогеноводороды большей частью ведут себя как неэлектролиты или слабые электролиты. При этом обычно наблюдается гораздо более резкое усиление ионизации по мере повышения атомного номера галоида, чем в водных растворах. Так, в пиридине константы диссоциации галогеноводородов имеют следующие значения: 3·10-9 — (НF), 4·10-6 (НСl), 1·10-4 (HBr), 3·10-3 — (НI).
| Галогеноводород | Теплота образования из элементов, кДж/моль | Ядерное расстояние, пм | Длина молекулярного диполя, пм | Тпл, °С | Ткип, °С | Растворимость в воде моль/л при 10 °С | Степень диссоциации в 0,1 н. растворе, % |
| HF | 272 | 92 | 36 | -83 | +19,5 | Ґ | 9,0 |
| HCl | 92 | 128 | 23 | -114 | -85 | 14 | 92.6 |
| HBr | 33 | 141 | 17 | -87 | -67 | 15 | 93,5 |
| HI | -25 | 162 | 9 | -51 | -35 | 12 | 95,0 |
По химическим свойствам НВr и НI очень похожи на хлористый водород. Подобно последнему в безводном состоянии они не действуют на большинство металлов, а в водных растворах дают очень сильные бромистоводородную и иодистоводородную кислоты. Соли первой носят название бромистых или бромидов, второй — иодистых или иодидов (а производные галогеноводородных кислот вообще — галогенидов). Растворимость бромидов и иодидов в большинстве случаев подобна растворимости соответствующих хлоридов. Возможность существования в виде отрицательно одновалентного иона установлена и для астата.
Существенное различие между НI, НВr и НСl наблюдается в их отношении к окислителям. Молекулярный кислород постепенно окисляет иодистоводородную кислоту уже при обычной температуре (причем под действием света реакция сильно ускоряется):
О2 + 4 НI = 2 Н2О + I2
Бромистоводородная кислота взаимодействует с ним гораздо медленнее, а соляная вовсе не окисляется молекулярным кислородом. Так как, однако, соляная кислота способна окисляться под действием MnО2 и т. п., из изложенного следует, что галоидоводороды (кроме НF) могут служить в качестве веществ, отнимающих кислород, т. е. в качестве восстановителей, причем наиболее активным в этом отношении является НI. Газообразный иодистый водород способен даже гореть в кислороде (с образованием Н2О и I2). Легкая окисляемость в растворах характерна и для производных отрицательно одновалентного астата.
Получение растворов иодистоводородной кислоты (вплоть до 50 %-ной концентрации) удобно вести, пропуская Н2S в водную суспензию иода. Реакция идет по схеме:
I2 + Н2S = 2 НI + S
Для предохранения водных растворов от окисления кислородом воздуха рекомендуется добавлять к ним небольшое количество красного фосфора (1 г/л), который, будучи практически нерастворимым в иодистоводородной кислоте, вместе с тем тотчас переводит образующийся при окислении свободный иод снова в НI.
Выделяющийся при частичном окислении иодистоводородной кислоты свободный иод не осаждается, а остается в растворе вследствие взаимодействия с избытком ионов I– по схеме: I– + I2 = I3– + 16,7 кДж/моль. Аналогично могут возникнуть ионы Вr3– и СI3–, а также ионы Г3– образованные разными галоидами (кроме фтора). Образующийся в растворе ион Г3– находится при этом в равновесии с продуктами своего распада: Г3–Ы Г– + Г2. Устойчивость ионов Г3–, зависит от природы галоида и характеризуется следующими значениями констант равновесия:
[Г3–]/[Г2]·[Г–] = K Г Сl Br I
K 0,2 16 700
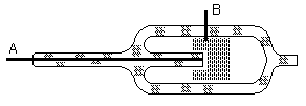
Рис.. Схема простейшего хемотрона.
Как видно из приведенных данных, по ряду С1-Вг-I устойчивость ионов Г3– быстро возрастает. Разбавление растворов и нагревание благоприятствуют смещению равновесий вправо, большая концентрация Г– — влево. Результатом существования подобных равновесий является более высокая растворимость свободных галоидов в растворах галогенидов по сравнению с чистой водой.Система 3 I–Ы I3– + 2 е- часто служит рабочей средой хемотронов — электрохимических установок для разностороннего оперирования со слабыми электрическими токами. Показанный на рис. простейший хемотрон представляет собой небольшой замкнутый сосуд, заполненный раствором КI с незначительной добавкой свободного иода (т. е. содержит много ионов I– и мало ионов I3-). Из двух впаянных платиновых электродов линейный (А) имеет малую рабочую поверхность, а сетчатый (Б) — большую. При включении тока в такой установке идут реакции:
3 I–- 2 е- = I3– — у анода и 2 е-+ I3– = 3 I– — у катода.
Если анодом является электрод А, а катодом — Б, то ионов I– около первого много (благодаря их высокой концентрации в растворе), ионов I3– около второго электрода тоже много (благодаря его большой поверхности), и ток свободно идет. Напротив, имеющийся около катода А небольшой запас ионов I3–, почти мгновенно исчерпывается, и ток практически прерывается. Рассматриваемая установка может, следовательно, служить выпрямителем слабых переменных токов низких частот, вообще же различные варианты хемотронов находят самое разнообразное техническое использование (например, в системах управления ракетными двигателями).
При рассмотрении кислородных соединений брома и иода, как и в случае хлора, удобно исходить из обратимой реакции
Г2 + Н2О Ы НГ + НОГ
равновесие которой при переходе от хлора к брому и затем иоду все более смещается влево.
В зависимости от природы галогенида, константы равновесия гидролиза имеют следующие значения:
[Н+]·[Г–]·[НОГ]/[Г2] = K 3·10-4 4·10-9 5·10-13
Г Cl Br I
В щелочной среде действительна иная трактовка гидролиза свободных галоидов, а именно по схеме:
Г2 + ОН–Ы НОГ + Г–
Растворы бромистоватистой (HOBr) и иодноватистой (HOI) кислот могут быть получены аналогично хлорноватистой кислоте. Обе кислоты являются неустойчивыми соединениями и сильными окислителями. По ряду HOCl-HOBr-HOI и устойчивость и окислительная активность уменьшается.
В том же направлении от хлора к иоду, ослабляется и кислотный характер соединений НОГ. Бромноватистая кислота является уже очень слабой, тогда как иодноватистая обладает амфотерными свойствами. Обе кислоты известны только в разбавленных растворах желтоватой или зеленоватой окраски со своеобразными запахами.
Вероятно, удобным путем получения бромноватистой кислоты могла бы быть реакция по схеме:
Ag2SO4 + Вr2 + Ва(ОН)2 = 2 АgВrЇ + ВаSO4Ї + 2 НОВг
Перегонку растворов НОВr (К = 2·10-9) можно производить только под уменьшенным давлением (ниже +30 °С), а НОI без разложения вообще не перегоняется. Обе кислоты известны лишь в растворах (НОВr — до 30 %-ной концентрации). Особенно неустойчивая иодноватистая кислота может быть несколько стабилизирована добавлением иода ( в результате равновесия НOI + I–Ы НOI2). Константа диссоциации НOI по кислотному типу (К = 2·10-11) даже меньше, чем по основному (3·10-10). Для реакции по уравнению
Н2О + Н2OI-Ы Н3О+ + НOI
было получено значение константы равновесия К = 3·10-2. Это значит, что при [Н3О+] = 1 (и отсутствии ионов I–) более трети всего растворенного количества НOI находится в форме ионов Н2OI+ (т. е. I+). С возможностью аналогичной основной диссоциации приходится считаться и у НОВr, и даже у НОСl.
Из солей обеих кислот в твердом состоянии были выделены только KOВr·3Н2О и кристаллогидраты NаОВr с 5 и 7 молекулами воды. Все эти светло-желтые соли очень неустойчивы, а при нагревании (или подкислении раствора) тотчас распадаются на соответствующие бромид и бромат.
43) Термическим разложением LiВгО3 при 200 °С был получен бромит лития — LiВrО3. Он представляет собой белый порошок, уже в присутствии следов воды разлагающийся по уравнению
3 LiВrО2 = LiВr + 2 LiВrО3
а при температуре плавления (225 °С) распадающийся на LiВr и O2. Аналогичные свойства характерны и для получаемого подобным же образом Ва(ВrО2)2.
44) При низких температурах (порядка -50 °С) бром окисляется озоном но реакции:
4 О3 + 3 Вr2 = 6 ВrО2
Образующийся диоксид брома (теплота образования из элементов — 54 кДж/моль) представляет собой светло-желтое твердое вещество, устойчивое лишь ниже -40 °С. Одним из продуктов её термического разложения в вакууме является коричневый гемиоксид брома (Вr2О), плавящийся при -17 °С (с разложением) и дающий с водой НОВr. Гемиоксид брома частично образуется также при действии брома на сухой оксид ртути или его взвесь в СС14. Он устойчив лишь ниже -40 °С. Аналогичный оксид иода известен только в форме оранжево-красного двойного соединения с пиридином — I2O·4С5Н5N.
Помимо окислительного распада, для HOBr и HOI очень характерны реакции по схеме:
3 НОГ = 2 НГ + НГО3
ведущие к образованию бромноватой (HBrO3) или иодноватой (HIO3) кислоты. Первая известна только в растворах, а вторая может быть выделена в виде легкорастворимых кристаллов. Обе кислоты бесцветны.
Бромноватая кислота очень похожа по свойствам на HClO3, тогда как и окислительные, и кислотные свойства иодноватой выражены значительно слабее. По рядуHClO3-HBrO3-HIO3 растворимость солей, как правило, уменьшается. Подобно хлоратам, броматы и иодаты в щелочных и нейтральных средах окислителями не являются.
Скорость реакции 3 НОГ = 2 НГ + НГО3 при переходе от хлора к брому и затем иоду быстро возрастает. Для брома было экспериментально установлено, что она максимальна при равной концентрации ОВr– и НОВr. Это позволяет предполагать активное участие в процессе молекул изобромноватистой кислоты — НВгО. И у брома, и у иода реакции протекают, вероятно, через промежуточное образование ионов ГО2–, однако аналогичные хлористой кислоте и хлоритам производные обоих элементов неизвестны. На приведенный выше основной процесс сильно налагается взаимодействие между НГ и НОГ. Поэтому общее уравнение разложения бромноватистой и иодноватистой кислот приближенно имеет вид:
5 НОГ = НГО3 + 2 Г2 + 2 Н2О.
Растворы бромноватой кислоты могут быть получены, в частности, по реакции:
5 АgВrО3 + 3 Вr2 + 3 Н2О = 5 АgВr + 6 НВгО3
Концентрировать их удается лишь до 50 %-ного содержания (т. е. приблизительно до состава НВrО3·7H2O). И окислительные, и кислотные свойства НВrО3 приблизительно таковы же, как у НСlO3. Для иона ВrО3- даются значения d(ВrО) = 178 пм и РОВгО = 112°.
Иодноватая кислота образуется, в частности, под действие хлора, на водную суспензию иода по реакции:
I2 + 5 Сl2 + 6 Н2О = 2 Н2O + 10 HCl
Поэтому при добавлении к раствору иодистой соли избытка хлорной воды появляющаяся вначале окраска иода затем вновь исчезает.
Для получения НIO3 (К = 0,2) обычно пользуются взаимодействием иода с крепкой азотной кислотой:
I2 + 10 НNО3 = 2 НIO3 + 10 NО2 + 4 H2O
Выделяющиеся окcиды азота удаляют пропусканием сквозь жидкость струи воздуха. Из сконцентрированного раствора при охлаждении осаждаются бесцветные кристаллы НIO3, плавящиеся при 110 °С (с переходом в НIO3·I2О5) и расплывающиеся на воздухе. Для молекулы НIO3 даются значения d(IO) = 180 пм (две связи) и 190 пм (одна связь), РOIO = 98°, а для иона IO3-, значения d(IO) = 182 пм и РOIO = 97°. В растворах иодноватой кислоты имеет место равновесие nНIO3 = (НIO3)3, где n = 2 или 3.
Растворимость производящихся от кислот НГО3 солей по ряду Сl-Br-I обычно уменьшается. Примером могут служить приводимые ниже данные (моль на литр Н2О при 20 °С):
NaClO3 | NaBrO3 | NaIO3 | KClO3 | KBrO3 | KIO3 | ||
| 9,2 | 2,3 | 0,46 | 0,58 | 0,41 | 0,38 |
В противоположность НСlO3 и НВrО3, для иодноватой кислоты, характерна совместная кристаллизация с её солями. Известны NаIO3·2HIO3, КIO3·НIO3, KIO3·2НIO3 и т. д. Получены были также некоторые продукты присоединения к иодатам иодноватого ангидрида, например КIO3·I2О5 (т. пл. 316 °С).
Подобные соли иногда рассматривают как производные “трииодноватой” кислоты — НI3O8. Доводом в пользу такой трактовки может служить возможность получения свободной НI3O8 как путем частичного термического разложения НIO3, так и путем её перекристаллизации из концентрированной НNО3. Однако “молекула” НI3O8 слагается из отдельных молекул НIO3 и I2O5, между иодными и кислородными атомами которых существует лишь сильное межмолекулярное взаимодействие.
Осторожным обезвоживанием HIO3 может быть получен белый порошок иодноватого ангидрида — I2O5. Он обладает сильными окислительными свойствами, а с водой вновь дает иодную кислоту.
В отличие от оксидов других галоидов, I2O5 является экзотермичным соединением (теплота образования 184 кДж/моль). Практически он может быть получен постепенным нагреванием НIO3 до 120 °С с последующим длительным выдерживанием при этой температуре. Кристаллы иодноватого ангидрида слагаются из молекул O2I-O-IO2 со значениями d(OI) = 177ё183 пм, РOIO = 93ё102° для концевых частей и d(IO) = 192ё195 пм, РIOI = 139° — для центральной части. Продажный препарат обычно имеет розоватый или желтоватый оттенок (обусловленный следами свободного иода). Продажный ангидрид постепенно разлагается на свету и очень гигроскопичен. Применяется он главным образом при газовом анализе для определения монооксида углерода (основанного на реакции I2O5 + 5 СО = 5 СО2 + I2).
При действии тлеющего разряда на смесь паров брома с избытком охлажденного кислорода образуется триоксид брома — ВrО3 (вероятно, в димерной форме — Вr2О6). Оксид этот (которому ранее приписывали формулу Вr2О6) представляет собой бесцветное кристаллическое вещество, устойчивое лишь ниже -70 °С. С водой он образует, по-видимому, две кислоты — НBrО3 и НВrO4, из которых последняя тотчас же разлагается на HBrО3 и кислород.
Вместе с тем взаимодействием Вr2 с избытком озона были получены Br3O8 и Вr2О5, но получить таким путем Вr2О6 не удалось. Вопрос о высших оксидах брома остается, таким образом неясным.
Соли бромной кислоты (HBrO4) образуются при окислении броматов фтором в щелочной среде:
NaBrO3 + F2 + 2 NaOH = 2 NaF + NaBrO4 + H2O
Сама кислота по силе близка к хлорной, но гораздо менее устойчива (известна только в растворе) и является более сильным окислителем. Её соли (перброматы) похожи по свойствам на перхлораты.
Иодная кислота (HIO4) может быть получена электролизом раствораHIO3 [по схеме H2O + HIO3 = H2(катод) + HIO4(анод)]. Выделяется она в виде бесцветного кристаллогидрата HIO4·2H2O. Кислотные свойства HIO4 выражены несравненно слабее, чем у HClO4, а окислительные, наоборот, гораздо более отчетливо. Большинство солей иодной кислоты (периодатов) малорастворимо в воде.
Несмотря на неоднократные попытки, бромную кислоту впервые удалось получить только в 1968 г. При обычных условиях её бесцветный раствор устойчив приблизительно до 6 М концентрации (55 %-ного содержания). Более крепкие растворы при хранении желтеют (вследствие восстановления НВrO4 до свободного брома). Как окислитель бромная кислота значительно сильнее хлорной, но окисляет она медленно (как и хлорная). Растворимость КВrО4, при комнатной температуре составляет около 0,2 М, т. е. несколько больше, чем у КСlO4. Ион ВrО4-, представляет собой тетраэдр с d(ВrО) = 161 пм. Пербромат калия термически устойчив до 280 °С (против 610 °С для КСlO4). Получен и пербромат аммония — NН4ВrO4.
Как кислота НIO4 (K = 3·10-2) слабее иодноватой. Наоборот, как окислитель она более активна, чем HIO3 (но менее, чем НOI). Весьма интересно отношение НIO4 к воде. При их взаимодействии в зависимости от условий может образоваться несколько соединений общей формулы (НIO4)n·(Н2О)m. Во всех таких соединениях водороды воды способны замещаться на металл так же, как и водород самой НIO4. В связи с этим соединения подобного типа обычно рассматривают как сложные кислоты и приписывают им следующие формулы: НIO4 (n=1, m=0), Н3IO5 (n=1, m=1), Н4I2O9 (n=2, m=1), Н5IO6 (n=1, m=2). Например, были получены К4I2O9 и следующие серебряные соли: оранжевая АgIO4, красная АgНIO5, черная Аg3IO5, зеленовато-желтая Аg2Н3IO6 и черная Аg5IO6.
При взаимодействии НIO4 с 65 %-ным олеумом образуется оранжевое твердое вещество. Судя по результатам анализа, оно представляет собой иодный ангидрид — I2O7. Свойства его пока не изучены. Двойным соединением I2O7·I2O5 является, вероятно, желтый продукт термического разложения Н5IO6 в вакууме при 110 °С.
Соли иодных кислот, как правило, труднорастворимы в воде. Некоторые из них весьма термически устойчивы (например, Nа5IO6 выдерживает без разложения нагревание до 800 °С). Получают периодаты обычно действием хлора в щелочной среде на соли иодноватой кислоты (например, по реакции
NaIO3 + 4 NаОН + Сl2 = Nа3Н2IO6 + 2 NаСl + Н2О
или же электролизом растворов солей HIO3.
Сообщалось, что термический распад Nа2Н3IO6 около 200 °С ведет к образованию Nа2IO4. Магнитные свойства препарата подтверждают как будто, что это вещество является производным шестивалентного иода. Оно устойчиво до 370 °С, а водой тотчас разлагается на иодат и периодат. Подобным же образом были получены некоторые другие соли, предположительно также являющиеся производными шестивалентного иода.
Кроме рассмотренных выше кислородных соединений брома и иода, известны еще некоторые. Из них наиболее интересны производные трёхвалентного иода, в которых он играет роль металла. Например, были получены устойчивый лишь ниже 0 °С желтый I(NО3)3, желтый IРO4, желто-зеленый I(ClO4)3·2Н2О и бесцветный I(СН3СОО)3. При электролизе последней соли иод выделяется на катоде, чем и доказывается его положительный заряд. Из аналогичных производных брома известен бесцветный Br(NO3)3.
Солеобразные производные одновалентных иода и брома очень неустойчивы сами по себе, но некоторые из них довольно устойчивы в виде двойных соединений с пиридином. Например, желтый INО3 разлагается уже выше -5 °С, тогда как бесцветный INО3·2С5H5N плавится при 138 °С без разложения. Сходные свойства имеют желтый ВrNО3 (т. пл. -42 °С) и бесцветный ВrNО3·2С5Н5N (т. пл. 80 °С). Известны также аналогичные нитратам по составу перхлораты и производящиеся от одновалентного иода соли некоторых органических кислот. Наиболее интересным из этих производных Вr+ является бромперхлорат, который был получен при -45 °С по реакции
Вr2 + 2 СlClO4 = Сl2 + 2 ВrСlO4
и представляет собой красную жидкость, еще не замерзающую при -78 °С и медленно разлагающуюся уже при -20 °С. Озонированием ВrNО3 был получен очень неустойчивый оранжевый ВrО2NО3.
Растворение смеси 2 I2+3 I2O5 (что эквивалентно 5 I2O3) в концентрированной Н2SO4 ведет к образованию желтых расплывающихся на воздухе кристаллов (IO)2SO4. При обработке дымящей Н2SО4 они белеют, по-видимому, вследствие перехода в (IO)НSО4. Обработка сульфатов иода водой сопровождается выделением I2 и желтого труднорастворимого порошка состава I2O4. Оксид этот, разлагающийся выше 100 °С на I2 и I2O5, следует рассматривать как основную иодноватокислую соль трехвалентного иода — (IO)IO3.
При обработке иода озоном образуется желтоватый расплывающийся на воздухе (с разложением) порошок состава I4O9. По-видимому, он представляет собой среднюю иодноватокислую соль трехвалентного иода — I(IO3)3. Выше 75 °С иодтрииодит разлагается с выделением иода.
Как видно из рассмотренного выше материала, аналогия брома и иода с хлором в их кислородных соединениях выражена уже далеко не столь полно, как в водородных: закономерный характер изменения свойств при переходе по ряду Cl-Br-I здесь ограничивается главным образом кислотами типов НОГ и НГО3 и их солями. О кислородных соединениях астата известно лишь, что они существуют, причем высшая степень окисления отвечает иону AtO3-, т. е. степени окисления +5.
Астат несколько менее летуч, чем иод, и из разбавленных азотнокислых растворов не отгоняется (в отличие от иода). Сероводородом в солянокислой среде он осаждается вместе с Вi2S3 и Sb2S3, а обработка осадка сернистым аммонием частично переводит At в раствор. Состав образующихся при этом его соединений не установлен. Самый концентрированный из подвергавшихся исследованиям раствор соединений астата был относительно него 10-8 М.
Наиболее сильными окислителями (в частности, НОС1) астат окисляется до иона АtО3–. Известна и другая, более низкая положительная валентность Аt, возникающая при его обработке менее сильными окислителями (Вr2, НNО3 и т. д.). По-видимому, в этих условиях образуется ион ОАt–. Установлена также возможность замещения астатом (Аt–) иода в его производных типа IХ·2С5Н5N (где Х = NО3 или СlO4). Раствор FеSO4 восстанавливает окисленные состояния Аt до элементарного. Действием Zn в кислой среде (или SnСl2 в щелочной) астат может быть далее восстановлен до иона Аt–. Последний легко вновь окисляется до элементарного Аt.
15
·
Подгруппа ванадия.
Члены этой подгруппы — ванадий, ниобий и тантал — похожи друг на друга приблизительно так же, как Сr, Mo и W.
Ванадий открыт в 1830 г., ниобий — в 1801 г., тантал — в 1802 г. Природный ванадий состоит из двух изотопов — 50V (0,2 %) и 51V (99,8 %), тогда как ниобий (93Nb) и тантал (181Ta) являются “чистыми” элементами.
Для аналога тантала — радиоактивного элемента № 105 были предложены названия “Ганий” (На) и “Нильсборий” (Ns). В 1970 г. сообщалось о синтезе его изотопа с массовым числом 260 и средней продолжительностью жизни атома около 2 с.
Ванадий довольно широко распространён в природе и составляет около 0,005% от общего числа атомов земной коры. Однако богатые месторождения его минералов встречаются редко. Помимо таких месторождений, важным источником сырья для промышленного получения ванадия являются некоторые железные руды, содержащие примеси соединений этого элемента.
При выветривании минералов земной коры, содержащих ванадий, соединения этого элемента отчасти удерживаются почвой, отчасти выносятся поверхностными водами в океан. Так, современные донные отложения Кольского залива и Каспийского моря содержат около 0,02% ванадия. Наличие его в некоторых железных рудах осадочного происхождения, нефти и каменном угле свидетельствует о большой биологической роли этого элемента для отдельных видов животных и растительных организмов минувших эпох. Некоторые современные растения и простейшие морские животные (асцидии, голотурии и др.) также избирательно извлекают ванадий из окружающей среды и накапливают его в своих организмах. Установлено, что ванадием богаты мухоморы. На организмы теплокровных животных растворимые соединения ванадия действуют как сильные яды.
Содержание ниобия (2·10-4 %) и тантала (2·10-5 %) в земной коре значительно меньше, чем ванадия. Встречаются они главным образом в виде минералов колумбита [M(NbO3)2] и танталита [М(ТаО3)2] (где М — Fe, Mn), которые обычно образуют смеси друг с другом. Важной рудой ниобия является сложный по составу минерал лопарит (содержащий около 11 % Nb2O5).
Технологическая переработка руд V, Nb и Ta довольно сложна. Для получения свободных элементов может быть использовано взаимодействие их оксидов с металлическим кальцием по схеме:
Э2О5 + 5 Са = 5 СаО + 2 Э
Реакции начинаются при нагревании исходных смесей и протекают с большим выделением тепла (для ванадия — 807 кДж/моль). Металл выделяется в виде ковких корольков.
Для промышленного получения ниобия и тантала основное значение имеет электролиз их расплавленных фторидов К2ЭF7 (содержащих растворённые оксиды Э2О5). Металлы выделяются в виде порошков, которые переводят в компактное состояние методами порошковой металлургии.
Ванадий, ниобий и тантал представляют собой не изменяющиеся на воздухе серые металлы, в чистом состоянии хорошо поддающиеся механической обработке. Их физические свойства:
| V | Nb | Ta | |
Плотность, г/см3 | 6,1 | 8,6 | 16,6 |
Температура плавления, °С | 1890 | 2470 | 3000 |
Температура кипения, °С | 3390 | 4840 | 5300 |
| Относительная электропроводность (Hg=1) | 4 | 5 | 6 |
В компактном состоянии все три металла весьма устойчивы по отношению к различным химическим воздействиям. Ванадий растворяется только в HF или в кислотах, являющихся одновременно сильными окислителями. Ниобий и тантал
нерастворимы во всех обычных кислотах и их смесях (царской водке и др.). Исключением является НF, сама по себе лишь медленно действующая на оба металла, но легко растворяющая их в присутствии сильных окислителей, например по реакции:
3 Та + 21 НF + 5 HNO3 = 3 H2[TaF7] + 5 NO + 10 H2O.
Растворы щелочей на рассматриваемые металлы почти не действуют, но в расплавленных щелочах они растворяются.
При переходе по ряду V-Nb-Ta металлы темнеют. Теплоты плавления составляют соответственно 21 (V), 27 (Nb) и 31 (Та) кДж/моль, а теплоты атомизации (при 25 °С) равны 514 (V), 723 (Nb) и 782 (Ta) кДж/моль.
Высокая химическая стойкость Nb и Ta обусловлена легко протекающим образованием на поверхности обоих металлов тончайшей, но очень плотной оксидной плёнки, которая делает их “пассивными”. В отсутствие комлексообразования эти плёнки защищают Nb и Ta при любых значениях рН среды.
В виде порошков V, Nb и Ta при нагревании соединяются с кислородом, галогенами, серой и азотом. Все три металла способны поглощать значительные количества водорода, однако определённые соединения при этом не образуются.
Растворимость водорода в металлах подгруппы ванадия довольно велика, однако компактные металлы хорошо поглощают его лишь после предварительной подготовки (путём нагревания в атмосфере Н2 и затем в вакууме) или если они являются катодами при электролизе. Поглощение водорода сопровождается ростом твёрдости и хрупкости металла. При повышении температуры растворимость водорода последовательно уменьшается. В индивидуальном состоянии были получены NbH2 (серый, устойчивый на воздухе) и VH2 (медленно разлагающийся на воздухе), тогда как TaH2 получить не удалось.
Основной областью применения ванадия является металлургия специальных сталей, которым он сообщает весьма ценные качества. Использование ниобия и тантала ещё сравнительно невелико, но имеет тенденцию к быстрому развитию.
Ежегодная мировая добыча ванадия составляет примерно 50 тыс. тонн, причём выплавляется главным образом не сам металл, а феррованадий (35 ё 80 % V). Введение в сталь небольших количеств ванадия (порядка 0,2 %) значительно увеличивает её упругость, прочность на истирание и сопротивление разрыву. Ванадиевая сталь применяется для изготовления автомобильных и авиационных моторов, осей, рессор и т. д. Алюминиевые стали с присадкой ванадия важны для конструирования гидросамолётов и глиссеров, так как они характеризуются высокой прочностью, эластичностью и устойчивостью по отношению к действию морской воды. Значительную техническую ценность имеют и некоторые другие сплавы ванадия (например, ванадиевая бронза). Соединения ванадия применяются главным образом в резиновой, стекольной и керамической промышленности. Они часто служат также хорошими катализаторами (преимущественно окислительных реакций).
Основной областью применения ниобия является его введение в состав сталей, предназначенных для изготовления сварных конструкций. Применение это основано на том, что Nb резко повышает прочность сварных швов. Феррониобий содержит обычно 30-75 % Nb. Ниобий не взаимодействует с некоторыми расплавленными металлами (щелочными, Sn, Pb и др.) и до 1100 °С — с ураном, что важно для атомной техники. Небольшая добавка ниобия сильно повышает твёрдость меди и её сплавов. Специальные сплавы с участием ниобия (а также тантала) применяются в реактивной технике, ядерных реакторах, газовых турбинах и т. д. Работа выхода электрона для ниобия (4,0 эв) самая низкая среди чистых тугоплавких металлов. Находящаяся в разбавленной серной кислоте ниобиевая пластинка пропускает электрический ток только тогда, когда она является катодом. Такая униполярная проводимость может быть использована для выпрямления переменного тока. Ежегодная мировая добыча ниобия исчисляется сотнями тонн.
Чрезвычайная устойчивость тантала по отношению к различным химическим воздействиям (например, ниже 150 °С на него практически не действуют ни сухие, ни влажные Cl2, Br2 и I2) наряду с высокой твёрдостью, ковкостью и тягучестью, делают этот металл особенно пригодным для изготовления различных ответственных частей заводской химической аппаратуры. Широкому развитию такого применения мешает лишь высокая цена тантала. Металл этот (а также и Nb) широко используется в радиотехнической промышленности и электровакуумной технике. Работа выхода электрона для тантала составляет 4,1 эв. Тонкие танталовые пластинки и проволоки являются важным вспомогательным материалом костной и пластической хирургии. Обусловлено это тем, что тантал, в противоположность другим металлам (кроме ниобия), совершенно не раздражает соприкасающуюся с ним живую ткань. В результате танталовые заплаты на черепе, сшивки костей и т. д. нисколько не вредят жизнедеятельности организма. Ежегодная мировая выработка тантала исчисляется сотнями тонн.
Наиболее типичны для ванадия и его аналогов производные пятивалентных элементов. Кроме того, известны соединения, отвечающие валентностям IV, III и II. При переходе по ряду V-Nb-Ta число таких соединений и их устойчивость уменьшается. Производные низших валентностей ниобия и тантала практического значения пока не имеют.
Оксиды пятивалентных элементов (Э2О5) образуются при прокализании мелко раздробленных металлов в токе кислорода. Из них V2O5 имеет явно выраженный кислотный характер, а у Nb2O5 и Ta2O5 он значительно ослабляется.
Красный ванадиевый ангидрид (V2O5) малорастворим в воде. Его жёлтый раствор содержит довольно слабую ванадиевую кислоту (HVO3). В щелочах V2O5 легко растворяется, образуя соответствующие ванадаты, из которых наиболее важен сравнительно малорастворимый ванадат аммония (NH4VO3), являющийся обычным реактивом ванадия.
Ванадиевый ангидрид удобно получать нагреванием NH4VO3 на воздухе. В мелко раздробленном состоянии он имеет оранжевый или жёлтый цвет. Расплавленный V2O5 (т. пл. 685 °С) проводит электрический ток, что заставляет предполагать наличие незначительной электролитической диссоциации по схеме:
V2O5Ы VO2+ + VO3-.
С водяным паром при 500-600 °С гемипентаоксид ванадия заметно летуч, что обусловлено, по-видимому, существованием равновесия по схеме:
V2O5(тв.) + 2 H2O(газ) Ы V2O3(OH)4(газ).
Насыщенный при обычных условиях водный раствор содержит около 0,04 % V2O5. Для него известны кристаллогидраты с 3, 2 и 1 молекулами воды, по составу отвечающие орто-, пиро- и мета-формам ванадиевой кислоты. Растворимость V2O5 в разбавленных сильных кислотах значительно выше, чем в воде, что указывает на проявление ванадиевым ангидридом заметных признаков амфотерности.
Для ванадиевой кислоты (К = 2·10-4) оба возможных направления электролитической диссоциации
VO3’ + H•Ы VO2OH Ы VO2• + OH’
по вероятности протекания они соизмеримы друг с другом. В сильнокислых растворах (с рН 2+, которые характеризуются отчётливо выраженными окислительными свойствами. Так, в сильнокислой среде хлористый водород медленно окисляется ими до свободного хлора. Реакция идёт по схеме:
2 VO2• + 2 HCl Ы 2 VO•• + Cl2 + 2 OH’.
Хорошо растворимы в воде только ванадаты немногих одновалентных металлов. Растворы их бесцветны или окрашены в желтоватый цвет. Чистый ванадат аммония бесцветен, но при нагревании выше 30 °С легко теряет часть аммиака и желтеет. Жёлтую окраску имеет также его раствор (растворимость 1:100 при обычных температурах). Ванадаты двух- и трёхвалентных металлов, как правило, малорастворимы в воде.
Жёлтый цвет растворов ванадатов аммония обусловлен, по-видимому, образованием в них ионов V3O9’’’ по схеме 3 VO3’Ы V3O9’’’ (K = [VO3’]2/ [V3O9’’’] (для константы равновесия этой реакции в нейтральной среде было найдено значение К = [ VO3’]3/[ V3O9 ’’’] = 3·10-6). В кислых средах для ванадия характерно образование солей типа М4[V6O17] или M2[V6O16] (так называемых гексаванадатов), большинство которых окрашено в цвета от золотисто-жёлтого до рубинового. Переход от обычных метаванадатов (с ионом VO3’ или V3O9’’’) к гексаванадатам соответствует схемам:
2 V3O9’’’ + 2 H•Ы H2O + V6O17’’’’ или 2 V3O9’’’+ 4 H•Ы 2 H2O + V6O16’’.
По другим данным, основной формой существования ванадиевой кислоты в умеренно кислых средах (рН = 1,5 ё 6,5) является H6V10O28. Для двух последних констант диссоциации этой кислоты (в 1 М растворе NaClO4) были найдены следующие значения: К5 = 2·10-4 и К6 = 8·10-7.
Замена кислой реакции на щелочную обуславливает образование анионов пиро- и ортованадиевой кислот по схемам:
2 V3O9’’’+ 6 OH’ = 3 H2O + 3 V2O7’’’’ и V2O7’’’’ + 2 OH’ = H2O + 2 VO4’’’.
Ортованадат натрия (Na3VO4) гидролитически разлагается водой на холоду до пированадата (Na4V2O7), а при кипячении — до метаванадата (NaVO3). При условии точной дозировки рН среды соответствующая серебряная соль может быть выделена во всех трёх формах и из слабокислых растворов:
| рН | 4.3-4,7 | 5,5-5,8 | 6,0-6,5 |
| Осаждается | AgVO3 | Ag4V2O7 | Ag3VO4 |
Мета-, пиро- и ортованадаты калия плавятся соответственно при 520, 910 и 1300 °С. Наличием подобных солей устанавливается сходство гидратных форм ванадиевой и фосфорной кислот, тогда как сильно выраженная у первой из них склонность к полимеризации в кислой среде сближает ванадий с хромом.
Из производных, отвечающих основной функции ванадиевой кислоты, известны красные твёрдые VO3NО3 и VО2ClO4, жёлтые VO(NO3)3 (т. пл. 2 °С) и VO(ClO4)3 (т. пл. 22 °С). Взаимодействием VOCl3 с раствором SO3 в SO2Cl2 был получен оксосульфат — V2O(SO4)4. Все эти соединения малоустойчивы. Напротив, жёлтые кристаллы VOPO4·2Н2O вполне устойчивы. Интересно, что насыщенный раствор этого соединения (растворимость около 1 вес. %) имеет тёмно-фиолетовый цвет.
При действии на раствор NH4VO3 сернистого аммония жидкость окрашивается в вишнёво-красный цвет вследствие образования тиосоли по суммарному уравнению:
NH4VO3 + 4 (NH4)2S + 3 H2O = (NH4)3VS4 + 6 NH4OH
Твёрдый тиованадат аммония представляет собой фиолетовые кристаллы, легкорастворимые в воде. Подобно аналогичным производным фосфора, в растворах тиованадаты подвергаются гидролизу с последовательным образованием ионов, промежуточных по составу между VS4’’’ и VO4’’’.
Выдерживание (NH4)2VS4 при 60 °С в токе сухого азота (не содержащего примеси кислорода) ведёт к распаду его на NH3, H2S и чёрный V2S5. Последний нерастворим в воде (но растворяется в щелочах) и легко окисляется кислородом. Выше 300 °С он переходит в V2S3. Описан также полисульфид состава VS5, при нагревании которого происходит последовательное отщепление серы, и состав меняется по ряду VS4 (300 °С) — VS2 (400 °С) — V2S3.
Бесцветные Nb2O5 и Ta2O5 тугоплавки и в воде почти нерастворимы. Отвечающие им соли — ниобаты и танталаты могут быть получены сплавлением соответствующего ангидрида со щелочью (или окислами металлов). В водных растворах они сильно гидролизованы. При подкислении этих растворов выделяются белые студенистые осадки переменного состава Э2О5·хН2О. Оба гидроксида растворимы не только в крепких растворах щелочей, но и в сильных кислотах, что указывает на их амфотерность.
Константы первой ступени кислотной и основной диссоциации гидроксидов ниобия и тантала составляют соответственно 4·10-8 и 3·10-15 (Nb) или 3·10-10 и 1·10-13 (Ta). Обезвоживание осадков Э2О5·хН2О нагреванием сопровождается (при потере последней гидратной воды) саморазогреванием массы, обусловленным выделением тепла при переходе оксида из аморфного в кристаллическое состояние (теплота кристаллизации). И Nb2O5, и Ta2O5 известны в двух модификациях (точки перехода 830 и 1360 °С). Высокотемпературные формы плавятся соответственно при 1490 и 1870 °С.
Прокаливание в токе водорода ведёт к восстановлению Nb2O5 до NbO2 (и затем до NbO), тогда как Ta2O5 водородом не восстанавливается. Из очень тесных смесей обоих ангидридов образуются две твёрдые фазы — состава Э2О5 и ЭО2, из которых первая богата танталом, а вторая — ниобием. Так как в 80%-ной серной кислоте растворима только вторая фаза, этим можно воспользоваться для частичного разделения обоих элементов.
Состав ниобатов и танталатов сильно зависит от условий их получения. При выделении из раствора наиболее характерны гекса-соли М8Э6О19·nH2O или пента-соли М7Э5О16·nH2O. Сухим путём были получены также некоторые орто-соли типов М3ЭО4 и М5ЭО5. Большинство ниобатов и танталатов малорастворимо в воде. Растворимые соли (главным образом, производные калия) подвергаются сильному гидролизу.
Из производных, отвечающих основной функции гидроксидов Э(ОН)5, лучше других изучены сульфаты и фосфаты. Для ниобия описаны оксосульфаты Nb2O4SO4, Nb2O3(SO4)2 и Nb2O(SO4)4, а для тантала даже нормальный сульфат Ta2(SO4)5. Известны также оксонитраты ЭO(NO3)3. Водой все эти бесцветные кристаллические вещества легко гидролизуются. Сухим путём были получены нерастворимые в воде оксофосфаты ЭОРО4 и нормальные фосфаты Э3(РО4)5 обоих элементов.
Для всех элементов рассматриваемой подгруппы характерно образование пероксидных солей, устойчивость которых по ряду V-Nb-Ta повышается. Производятся они главным образом от орто- (Н3ЭО4) или мета- (НЭО3) гидратов путём замены части или всех атомов -О- на перекисные группы -О-О-. Так, при действии H2O2 на V2O5 в концентрированной щелочной среде образуются сине-фиолетовые ионы VO83-, а в близкой к нейтральной разбавленной — жёлтые ионы VO63-. В кислой среде образуются красный пероксидный катион VO3+, а при очень высокой кислотности происходит восстановление ванадия до синего VO2+. Свободные надкислоты ванадия не выделены, но некоторые надванадаты (например, Na3VO8) были получены и в твёрдом состоянии.
При действии H2O2 на водные растворы сплавов Nb2O5 и Ta2O5 с KOH образуются бесцветные пероксидные соли состава К3ЭО8. Аналогичные соли выделены и для некоторых других катионов. Действием на растворы надниобатов и надтанталатов разбавленной H2SO4 могут быть получены (в виде кристаллогидратов) и свободные надкислоты. Обе они отвечают мета-форме и довольно устойчивы. Например, лимонно-жёлтый кристаллогидрат HNbO4·nH2O разлагается разбавленной серной кислотой (с отщеплением H2O2) лишь при нагревании, а бесцветный кристаллогидрат HTaO4·nH2O выдерживает нагревание до 100 °С без разложения.
Галогениды для пятивалентных элементов не характерны (известен только VF5). Для Nb и Ta могут быть получены все возможные пентагалогениды ЭГ5. Они представляют собой легкоплавкие и легколетучие кристаллические вещества. Фториды и хлориды бесцветны, тогда как бромиды и иодиды имеют различные цвета — от жёлтого до чёрного. Водой все пентагалогениды разлагаются с выделением осадка соответственно ниобиевой или танталовой кислоты (Э2О5·хН2О). Для фторидов характерна тенденция к комплексообразованию, причём большинство производящихся от них комплексных соединений отвечает типу М2[ЭГ7], где М — одновалентный металл.
Фторид пятивалентного ванадия может быть получен взаимодействием элементов при 300 °С (теплота образования 1471 кДж/моль) и представляет собой бесцветное кристаллическое вещество (т. пл. 19, т. кип. 48 °С). Молекула VF5 имеет форму правильной треугольной бипирамиды, а связь VF в ней характеризуется длиной 171 пм и энергией 477 кДж/моль. В жидком состоянии, по-видимому, имеет место частичная ионизация ванадийпентафторида по схеме:
2 VF5Ы VF4+ + VF6-.
Со многими веществами (например с PСl3) он реагирует весьма бурно, а водой полностью гидролизуется.
Молярная растворимость VF5 в жидком HF равна приблизительно 1:15 причём тенденция к образованию HVF6 выражена слабо и сама комплексная кислота не выделена, но получены некоторые производящиеся от неё соли. По отношению к нагреванию они не особенно устойчивы. Так, K[VF6] распадается на KF и VF5 уже при 330 °С. Были получены также твёрдый при обычных условиях 2XeF6·VF5 (давление пара 5 мм. рт. ст.) и жидкий 2XeOF4·VF5 (т. пл. -37 °С). Интересно, что получить аналогичные продукты присоединения с молекулярным соотношением 1:1 не удалось.
Плотности паров пентагалогенидов отвечают простым молекулам ЭГ5. Последние имеют структуру тригональной бипирамиды с атомом Э в центре [d(NbГ) = 188 (F), 228 (Cl), 246 (Br) и d(TaГ) = 186 (F), 227 (Cl), 245 (Br). для энергий связей даются следующие значения (кДж/моль): 410 (NbCl), 426 (TaCl), 343 (NbBr), 360 (TaBr). Теплоты образования из элементов, температуры плавления и кипения пентагалогенидов Nb и Ta сопоставлены ниже:
| Теплота образования (кДж/моль) | Температура плавления, °С | Температура кипения, °С | |
NbF5 | 1810 | 79 | 234 |
NbCl5 | 800 | 205 | 248 |
NbBr5 | 556 | 268 | 362 |
NbI5 | (426) | 320 | разл. |
TaF5 | 1900 | 97 | 230 |
TaCl5 | 857 | 217 | 234 |
TaBr5 | 598 | 280 | 349 |
TaI5 | (489) | 496 | 543 |
Пентафториды ниобия и тантала склонны к переохлаждению. В их расплавах имеет место незначительная (
2 ЭF5Ы ЭF4+ + ЭF6-.
Взаимодействием NbF5 с жидким аммиаком был получен блестящий жёлтый аммиакат NbF5·2NH3. Пятихлористый ниобий известен в двух формах — белой и жёлтой (точка перехода 183 °С). Пентагалогениды довольно хорошо растворимы в эфире и способны образовывать с ним кристаллоэфираты ЭГ5·(С2Н5)2О. Интересно, что по ряду Cl-Br-I растворимость понижается (обычно для неорганических галогенидов и органических растворителей наблюдается обратное). Желтоватый хлорофторид NbCl4F способен существовать в ионной и молекулярной форме. Известен и бесцветный TaCl4F (т. пл. 214 °С). В твёрдом состоянии он тетрамерен.
Для обоих элементов известны также комплексы типов MI[ЭF6] и MII[ЭF6]2, а для тантала получены и комплексы типа M3[TaF8], например K3[TaF8] (т. пл. 780 °С). Свободные фторониобиевая и фторотанталовая кислоты известны в виде бесцветных, плавящихся около 15 °С кристаллогидратов Н[ЭF6]·6Н2О. Из всех этих комплексных производных наибольшее значение имеет малорастворимый в холодной воде бесцветный K2[TaF7] (т. пл. 775 °С), легко образующийся при растворении Ta2O5 в содержащей KF плавиковой кислоте. Соль эта, выделяющаяся без кристаллизационной воды, гораздо лучше растворима при нагревании, чем на холоду, и поэтому может быть легко очищена перекристаллизацией (из растворов, во избежание гидролиза подкисленных HF). Этим обычно и пользуются для очистки тантала от примесей, в частности для отделения его от ниобия, который в отсутствии большого избытка HF образует довольно хорошо растворимый оксофторидный комплекс K2[NbOF6].
Для пентафторидов ниобия и тантала известны жёлтые двойные соединения с ксенондифторидом типов XeF2·ЭF5 и XeF2·2ЭF5. Они представляют собой легкоплавкие и малоустойчивые кристаллические вещества, вероятно, сходные по строению с аналогичными производными SbF5.
Азотные производные пятивалентных элементов характерны главным образом для тантала. Красный Та3N5 образуется в результате взаимодействия Ta2O5 с NH3 при 900 °С. Если вести процесс при 800°С, то образуется жёлто-зелёный TaON. Известен и NbON. Аналогичный оксинитрид ванадия был получен по схемам:
VOCl3 + ClN3 = Cl2 + VOCl2N3 и VOCl2N3 = N2 + Cl2 + VON.
Синтезированы также нитрилхлориды ниобия и тантала, по составу аналогичные фосфонитрилхлоридам, но представляющие собой твёрдые кристаллические вещества. Жёлто-коричневый NNbCl2 отщепляет хлор около 450 °С, а жёлто-зелёный NTaCl2 — лишь при значительно более сильном нагревании. Из нитрилфторидов был получен NNbF2. Известен и нитрилхлорид состава Ta2N3Cl. Нитрилхлорид ванадия синтезирован по схемам:
VСl5 + ClN3 = Cl2 + VCl4N3 и VCl4N3 = N2 + Cl3VNCl
(он может быть получен и прямым взаимодействием VN с Cl2 при 130 °С). В отличие от полимерных нитрилгалогенидов тантала и ниобия это соединение (т. пл. 132 °С) мономерно и легко возгоняется. Для всех элементов подгруппы ванадия описаны двойные нитриды Li7ЭN4 (а для ванадия, кроме того, Li7VP4 и Li7VAs4).
Производные низших валентностей из рассматриваемых элементов более или менее характерны лишь для ванадия, Его тёмно-синий диоксид (VO2) имеет амфотерный характер (с преобладанием основных свойств над кислотными), а оба низших оксида — чёрные V2O3 и VO — обладают лишь основными свойствами. Соли этих оксидов и различных кислот имеют в растворах следующие характерные окраски: VO2 — голубую, V2O3 — зелёную и VO — фиолетовую. В кислой среде наиболее устойчивы производные четырёхвалентного ванадия, в щелочной — пятивалентного.
Обусловленное понижением валентности ванадия последовательное изменение окраски наглядно выявляется при действии цинка на солянокислый раствор ванадата аммония. Конечным продуктом восстановления в этом случае является V2+, тогда как Sn2+восстанавливает V5+ лишь до V3+, а I-до V4+.
Пятивалентный ниобий восстанавливается цинком в кислой среде до Nb+3, тогда как Та+5 совсем не восстанавливается.
Отвечающий четырёхвалентному состоянию синий оксид (VO2) может быть получен осторожным восстановлением V2O5 (например прокаливанием с избытком щавелевой кислоты). Сине-чёрный NbO2 (т. пл. 2080 °С) образуется в результате восстановления Nb2O5 водородом при 1200 °С. Для получения коричнево-черного TaO2 требуется очень энергичное восстановление Ta2O5 (например, магнием при высоких температурах). При нагревании на воздухе диоксиды легко переходят в соответствующие ангидриды Э2О5.
Для ванадия довольно характерны продукты частичного восстановления ванадатов приблизительно состава MxV2O5 (где 0 4, Cu, Ag, Pb). Эти “ванадиевые бронзы” по некоторым свойствам похожи на аналогичные соединения вольфрама. Ещё более сходны с последними “ниобиевые бронзы” типа МхNbO3 (где М — Na, K, Sr, Ba). Есть указание на существование “танталовых бронз” типа ВахТаО3.
Гидроксид четырёхвалентного ванадия отвечает формуле VO(OH)2. Он имеет розовый цвет, амфотерен и труднорастворим в воде (ПР = 2·10-22). Образующиеся при взаимодействии VO2 (т. пл. 1545 °С) со щелочами жёлтые или коричневые соли носят название ванадатов и обычно производятся от изополикислоты состава H2V4O9 (т. е. Н2О·4VO2). Легкорастворимые ванадаты калия и натрия кристаллизуются по типу М4[V4O9]·7Н2О. Мета- и пированадаты натрия были получены сухим путём (длительным нагреванием в вакууме) по реакциям:
2 NaVO3 + 2 NaN3 = 3 N2 + 2 Na2VO3 и V2O5 + 2 NaN3 = 3 N2 + Na2V2O5.
Ванадаты двух- и трёхвалентных металлов в воде практически не растворимы. Получают их обычно совместным прокаливанием VO2 и оксидов соответствующих металлов в вакууме.
Соли, образуемые диоксидом ванадия с кислотами, производятся от катиона VO2+ (ванадила). Они вполне устойчивы в кислых средах (даже при нагревании). Из них VOCl2 может быть проще всего получен растворением V2O5 в крепкой соляной кислоте. В твёрдом состоянии хлористый ванадил имеет зелёную окраску. Он весьма гигроскопичен и легко растворяется в воде с синим или бурым (в зависимости от условий) окрашиванием раствора. С синим окрашиванием растворяется в воде также буро-чёрный VOBr2. Аналогичный иодид получен в виде коричневого кристаллогидрата 2VOI2·5H2O, легкорастворимого в воде. То же относится к синему кристаллогидрату VOSO4·3Н2О (тогда как безводный сульфат ванадила имеет зелёный цвет и в воде практически нерастворим). С сульфатами некоторых других металлов VOSO4 образует двойные соли, главным образом типов M2SO4·2VOSO4 и M2SO4·VOSO4. И те и другие обычно выделяются с кристаллизационной водой. Возможно, что в качестве соли ванадила [VO(VO3)2·2H2O] следует рассматривать и довольно характерный для ванадия чёрный промежуточный гидроксид V3O5(OH)4. Чёрный амид ванадила [VO(NH2)2] уже при слабом нагревании переходит в имид [VO(NH)] и затем в нитрид [(VO)3N2].
Четырёххлористый ванадий может быть получен взаимодействием элементов около 200 °С. Он представляет собой тяжёлую красно-бурую жидкость (т. пл. -20, т. кип. 153 °С). Плотность его пара отвечает формуле VСl4. Молекула эта имеет структуру тетраэдра с атомом ванадия в центре [d(VCl) = 214 пм]. Аналогична структура и устойчивой лишь ниже -45 °С молекулы VВr4 [d(VBr) = 230 пм]. Для растворов ванадийтетрахлорида в CСl4 установлено наличие равновесия между простыми и димерными молекулами (частично характеризуемого соотношением [VCl4]2/[V2Cl8] = 2·10-2 при -24 °С). При нагревании VСl4 медленно распадается на VСl3 и хлор, а при взаимодействии с водой гидролизуется по уравнению:
VСl4 + H2O = VOCl2 + 2 HСl.
Производным зелёного VOCl2 является комплексная соль состава Cs3VOCl5.
Пропускание паров VСl4 над нагретыми до 400 °С хлоридами K, Rb и Cs ведёт к образованию продуктов присоединения типа M2VCl6, окрашенных соответственно в коричневый, розово-красный и фиолетовый цвет. Действием хлора на смесь VСl4 и S2Cl2 могут быть получены кристаллы двойного соединения VCl4·SCl4 (т. пл. 32 °С). При взаимодействии VСl4 с жидким аммиаком осаждается зелёновато-коричневый хлорид VCl(NH2)3, а при взаимодействии с NO образуются легко возгоняющиеся твёрдые вещества состава VCl4NO, V2Cl7NO, V2Cl8(NO)5. Вместе с тем взаимодействием VСl4 с NO в бензоле был получен коричневый невозгоняющийся полимер [V(NO)3Cl2]n.
Длительным нагреванием VСl4 с безводной HF может быть получен коричневый порошок VF4. При нагревании его выше 100 °С происходит дисмутация на VF5 и VF3. Ванадийтетрафторид гигроскопичен, хорошо растворим в воде и легко гидролизуется с образованием синего (в безводном состоянии жёлтого) фтороксида VOF2. Последний с фторидами ряда металлов даёт синие двойные соединения, главным образом типа M2[VOF4·H2O]. Известны и безводные соли типа K2[VOF4] и (NH4)3[VOF5]. Сухим путём были получены также розовато-жёлтые соли типа M2VF6 (где M = K, Rb, Cs).
Фиолетово-чёрный четырёххлористый ниобий может быть получен по схеме:
4 NbCl5 + Nb = 5 NbCl4
при 400 °С он начинает возгоняться около 275 °С, а выше 300 °С (при отсутствии избытка NbCl5) происходит его дисмутация по схеме:
2 NbCl4 = NbCl5 + NbCl3.
В небольшом количестве воды или в разбавленных кислотах NbCl4 растворяется с синим окрашиванием жидкости. Такие растворы характеризуются очень сильными восстановительными свойствами. Аналогично хлориду могут быть получены сходные с ним по свойствам чёрный NbF4 и коричневый NbBr4. Длительным нагреванием NbI5 до 270 °С в вакууме был получен серый NbI4. При 503 °С он плавится и с отщеплением части иода переходит в Nb2I8. Известны также бромид и хлорид аналогичного состава. При сплавлении NbCl4 с хлоридами щелочных металлов образуются нестойкие соединения типа M2NbCl6. По ряду Cs®Na их термическая устойчивость уменьшается.
Зеленовато-чёрный TaCl4 может быть получен при 600 °С по схеме:
4 TaCl5 + Ta = 5 TaCl4
в отсутствие избытка TaCl5 выше 280 °С наступает дисмутация по схеме:
2 TaCl4 = ТаСl3 + ТаСl5
(тогда как при 210 °С идёт обратная реакция). Четырёххлористый тантал является ещё более сильным восстановителем, чем NbСl4. Так, при 320 °С протекает реакция по схеме:
ТаСl4 + NbСl5 = ТаСl5 + NbCl4.
С хлоридами Cs, Rb, K танталтетрахлорид способен образовывать лиловые комплексные соли типа M2ТаСl5. Получен и тёмно-серый ТаI4. Оксохлориды ТаOСl2 и NbOCl2 были синтезированы сухим путём (нагреванием смесей Э, Э2О5 и ЭСl5 в запаянных трубках). Известен и чёрный NbOI2.
Сульфиды ЭS2 ниобия и тантала могут быть получены прямым взаимодействием элементов или нагреванием металлов в токе сухого сероводорода. Лучше изученный TaS2 представляет собой чёрный порошок, весьма термически устойчивый (в отсутствие воздуха) и нерастворимый ни в соляной кислоте, ни в растворах едкого натра. Известны и кристаллические фазы составов NbSe2, NbTe2, NbSe3, TaSe2, TaTe2, TaS3, TaSe2, TaTe4. Тёмно-серый сульфид трёхвалентного ванадия является фазой переменного состава (с областью гомогенности на интервале от VS1,17 до VS1,53).
Чёрный оксид трёхвалентного ванадия (V2O3) может быть получен восстановлением V2O5 водородом при 700 °С. Он медленно взаимодействует с кислотами, образуя соли, которые являются очень сильными восстановителями. При действии на их растворы щелочей выпадает зелёный осадок V(OH)3, чрезвычайно легко окисляющийся на воздухе.
Растворением V2O3 (т. пл. 1970 °С) в плавиковой кислоте и упариванием раствора может быть получен тёмно-зелёный VF3·3H2O. С фторидами ряда одновалентных (и двухвалентных) металлов VF3 образует комплексные соединения типов M2VF5 (обычно выделяющиеся с кристаллизационной водой) и M3VF6. Примером может служить бледно-зелёный K3VF6 (т. пл. 1020 °С). Безводный VF3 удобно получать термическим разложением (NH4)3VF6. Он имеет зеленовато-жёлтую окраску, нерастворим в обычных растворителях и плавится лишь около 1400 °С. Известен и оксофторид VOF.
Ванадийтрихлорид может быть получен разложением VСl4 при нагревании. Он представляет собой фиолетовые нелетучие кристаллы, легко растворимые в воде с зелёным окрашиванием раствора. При концентрировании последнего (в отсутствие кислорода воздуха) выделяется зелёный гигроскопичный кристаллогидрат VCl3·6H2O. Аналогичные ванадийтрихлориду чёрные бромид и иодид в общем похожи на него по свойствам, но отличаются меньшей устойчивостью. Образование комплексов с галогенидами других металлов для рассматриваемых соединений не характерно, но некоторые производные этого типа известны. Примером могут служить красные соли M2VCl5·H2O (где M — K, Rb, Cs, NH4) и K3VCl6 (т. пл. 744 °С). Интересны изменения цвета K2VCl5·nH2O в зависимости от величины n: фиолетовой (0), красный (1), зелёный (4). При взаимодействии VСl3 с аммиаком образуется V(NH2)Cl2, который около 300 °С переходит в V(NH)Cl и затем в VN. В жидком аммиаке может быть получен красно-коричневый [V(NH3)6]Cl3.
Протекающей при 300 °С дисмутацией по схеме:
2 VOCl2 = VOCl3 + VOCl
(или длительным нагреванием смеси V2O3 с VСl3 в запаянной трубке) может быть получен оксохлорид трёхвалентного ванадия. Он представляет собой коричневое твёрдое вещество, почти нерастворимое в воде, щелочах и кислотах. Термический распад VOCl наступает лишь около 800 °С. Известен и фиолетовый VOBr.
Из сернокислых производных для трёхвалентного ванадия наиболее характерны зелёная комплексная кислота H[V(SO4)2]·nH2O (где n = 6) и её соли, главным образом типа M[V(SO4)2]·12H2O. Они большей частью окрашены в различные оттенки фиолетового цвета, но дают зелёные растворы. При достаточно высоких концентрациях растворы эти по отношению к кислороду воздуха сравнительно устойчивы и окисляются им лишь медленно. Безводный V2(SO4)3 имеет жёлтый цвет и очень медленно растворяется в воде. Около 400 °С в вакууме он разлагается по схеме:
V2(SO4)3 = SO2 + 2 VOSO4.
Производные, отвечающие кислотной функции V(OH)3, получены сухим путём — сплавлением V2O3 с окислами наиболее активных металлов. Примерами могут служить LiVO2 и NaVO2, представляющие собой нерастворимые в воде чёрные порошки.
Нерастворимый в воде и разбавленных кислотах коричневый NbCl3 может быть получен восстановлением NbCl5 водородом при 400 °С. Близки к нему по свойствам чёрные NbBr3 и NbI3. Тёмно-синий NbF3 был получен нагреванием насыщенного водородом ниобия в струе H2 + HF. Он устойчив к действию и сильных кислот, и щелочей.
Трёхфтористый тантал и по способу получения, и по свойствам похож на NbF3. Зелёный TaCl3 может быть получен дисмутацией TaCl4. В отличие от нерастворимого NbCl3, с водой он образует зелёный раствор, обладающий сильными восстановительными свойствами. Щёлочи выделяют из этого раствора зелёный осадок Ta(OH)3, который при нагревании окисляется водой (с выделением водорода). При сплавлении TaCl3 с хлоридами Cs, Rb, K образуются красные комплексные соли типа M2TaCl5, характеризующиеся температурами плавления 710, 642 и 560 °С. Серо-зелёный TaBr3 был получен восстановлением TaBr5 водородом при 700 °С.
Прокаливанием мелкораздробленных V, Nb и Ta в токе азота могут быть получены их серые нитриды общей формулы ЭN. Все они устойчивы по отношению к воде и весьма тугоплавки (VN плавится при 2050 °С, NbN — при 2300 °С, а TaN — лишь при 3090 °С). Известны и фосфиды аналогичного состава ЭP. Были описаны также менее характерные для рассматриваемых элементов кристаллические фазы иных составов — V3N, Nb2N, Nb3N4, Ta2N, Ta4N5, Ta5N6, VP2, V3P2, V2P, V3P, NbP2, TaP2. Для нитрида VN характерна необычайно высокая твёрдость.
Для формально трёхвалентных ниобия и тантала установлено существование селенидов Э2Se3. Известны также кристаллические фазы состава Nb3Э (где Э — S, Se, Te).
Отвечающий двухвалентному ванадию чёрный оксид VO образуется при нагревании V2O5 до 1700 °С в токе водорода. При неизменности кристаллической структуры [типа NaCl с d(VO) = 205 пм] состав его может довольно сильно отклоняться от строгого соответствия формуле VO (в пределах VO0,85- VO1,25). Оксид ванадия (II) довольно хорошо проводит электрический ток. Он нерастворим в воде, но растворяется в разбавленных кислотах, образуя соответствующие соли окрашенного в фиолетовый цвет катиона V2+. Последние являются исключительно сильными восстановителями и при отсутствии окислителей постепенно выделяют из воды газообразный водород. Действием щелочей на их растворы может быть получен серо-фиолетовый осадок V(OH)2, не выделенный, однако, в чистом состоянии из-за его чрезвычайно лёгкой окисляемости.
Ванадийдихлорид VСl2 может быть получен протекающей выше 500 °С дисмутацией по схеме:
2 VCl3 = VCl4 + VСl2.
Его зелёные кристаллы плавятся лишь около 1350 °С, а фиолетовый водный раствор быстро зеленеет вследствие окисления V2+ до V3+. Восстановительные свойства VСl2 выражены даже сильнее, чем у CrCl2. Сходные свойства имеет коричневый бромид VBr2 и красный иодид VI2. Первый из них используется иногда в фотографии (как быстродействующий проявитель), второй — для получения очень чистого ванадия (термическим разложением при 1400 °С). Бледно-зелёный VF2 был выделен в виде сине-фиолетового кристаллогидрата VF2·4H2O. При сплавлении VСl2 с KСl образуются KVCl3 (т. пл. 946 °С) и менее устойчивый K2VCl4. Для галогенидов VГ2 известны довольно устойчивые аммиакаты.
Сульфат двухвалентного ванадия образуется при восстановлении металлическим цинком (или электролитическим путём) сернокислых растворов соединений ванадия. При принятии особых мер предосторожности против окисления он может быть выделен в виде фиолетового кристаллогидрата VSO4·7H2O. С сернокислыми солями некоторых одновалентных металлов VSO4 образует фиолетовые комплексные соли типа M2[V(SO4)2]·6Н2О. Последние сравнительно труднорастворимы и более устойчивы, чем сам сульфат двухвалентного ванадия.
Чёрный VS может быть получен взаимодействием элементов при 1000 °С. Кристаллы его способны без изменения структуры включать некоторый избыток серы (до состава VS1,17). То же самое характерно для NbS и NbSe. Известны также NbTe и TaSe.
Низшие оксиды ниобия и тантала могут быть получены по схеме:
Э2О5 + 3 С = 3 СО + 2 ЭО
при 1100 °С в вакууме. Лучше изученный серый NbO (т. пл. 1935 °С) имеет металлический вид и довольно хорошо проводит электрический ток.
Коричнево-чёрный NbCl2 может быть получен действием паров NbCl5 (в токе аргона) на нагретый до 700 °С металлический ниобий. Ниобийдихлорид устойчив на воздухе и нерастворим в воде. Восстановлением NbI3 водородом при 300 °С был получен и чёрный NbI2. В отличие от хлорида он медленно разлагается водой. Известен и NbBr2 (который был получен восстановлением NbBr5 водородом в электрическом разряде).
Тёмно-зелёный нелетучий TaCl2 может быть получен дисмутацией TaCl3 при 440 °С по уравнению:
3 TaCl3 = TaCl5 + 2 TaCl2.
Несмотря на то, что в воде TaCl2 практически нерастворим, он уже на холоду постепенно окисляется ею с выделением водорода.
И для тантала, и для ниобия характерна своеобразная атомная группировка Э6Cl12 [d(NbNb) = 285, d(TaTa) = 288, d(NbCl) = 241, d(TaCl) = 244 пм]. Эту группировку содержат зелёные кристаллы, выделяющиеся из раствора в HCl продукта восстановления ЭCl5 металлическим свинцом при 600 °С. Состав этих кристаллов — Э6Cl12·2HCl·7H2O или [Э6Cl12]Cl2·7H2O — пока не вполне ясен. В последнем случае они должны содержать атомы Э разных степеней окисления.
При сопоставлении элементов подгруппы ванадия с фосфором и азотом наблюдается резкое расхождение свойств производных низших валентностей и закономерный ход изменения характера высших оксидов. Действительно, при переходе по ряду N2O5, P2O5, V2O5, Nb2O5, Ta2O5 кислотный характер оксидов весьма последовательно ослабляется. Напротив, очень похожие на N и P в производных низших валентностей элементы подгруппы мышьяка уже не дают закономерного изменения химического характера высших оксидов при переходе от N к Bi. Хорошей иллюстрацией изложенного может служить приводимое ниже сопоставление теплот образования оксидов Э2О5 из элементов (кДж/моль):
| Sb | As | P | N | P | V | Nb | Ta |
| 1007 | 928 | 1492 | 42 | 1492 | 1555 | 1898 | 2044 |
Подгруппа галлия
Содержаниекаждого из членов данной подгруппы в земной коре по ряду галий (4·10-4 %) — индий (2·10-6 %) — таллий (8·10-7 %) уменьшается. Все три элемента чрезвычайно распылены, и нахождение в виде определённых минералов для них не характерно. Напротив, незначительные примеси их соединений содержат руды многих металлов. Получают Ga In и Tl из отходов при переработке подобных руд.
Все три члена рассматриваемой подгруппы открыты при помощи спектроскопа: таллий — в 1861 г., индий — в 1863 г. и галий — в 1875 г. Последний из них за 4 года до открытия был предстазан и описан Д. И. Менделеевым. Природный галлий слагается из изотопов с массовыми числами 69 (60,2 %) и 71 (39,8); нидий — 113 (4,3) и 115 (95,7); таллий — 203 (29,5) и 205 (70,5 %).
Для галлия известен редкий минералл галлит (СuGaS2). Следы этого элемента постоянно содержатся в цинковых рудах. Значительно большие его количества (до 1,5 %) были обнаружены в золе некоторых каменных углей. Однако основным сырьём для промышленного получения галлия служат бокситы, обычно содержащие незначительные его примеси (до 0,1 %). Извлекается он электролизом из щелочных жидкостей, являющихся промежуточным продуктом перепаботки природных бокситов на технический глинозём. Размеры ежегодной мировой выработки галлия исчисляются пока немногими тоннами, но могут быть значительно увеличины.
Индий получают главным образом в качестве побочного продукта при комплексной переработке сернистых руд Zn, Pb и Cu. Его ежегодная выработка составляет несколько десятков тонн.
Галлий концентрируется главным образом в пирите (FeS2). Поэтому шламы сернокислотного производства являются хорошим сырьём для получения этого элемента. Ежегодная мировая выработка таллия меньше, чем индия, но также исчисляется десятками тонн.
В свободном состоянии галий, индий и талий представляют собой серебристо-белые металлы. Их важнейшие константы сопоставлены ниже:
| Ga | In | Tl | |
Плотность, г/см3 | 5,9 | 7,3 | 11,9 |
Температура плавления, °С | 30 | 157 | 304 |
Температура кипения, °С | 2200 | 2020 | 1475 |
| Электропроводность (Hg = 1) | 2 | 11 | 6 |
По твёрдости галлий близок к свинцу, In и Tl — ещё мягче.
Для выделения Ga, In и Tl в свободном состоянии применяется или электролиз растворов их солей, или прокаливание оксидов в токе водорода. В парах металлы одноатомны.
Кристаллическая решётка галлия образована не отдельными атомами (как обычно для металлов), а двухатомными молекулами (d(GaGa) = 248 пм). Она представляет таким образом, интересный случай сосуществования молекулярной и металлической структур. Молекулы Ga2 сохраняются и в жидком галлии, плотность которого (6,1 г/см3) больше плотности твёрдого металла (аналогия с водой и висмутом). Повышение давления сопровождается снижением температуры плавления галлия. При высоких давлениях, помимо обычной модификации Ga(I), установлено существование двух других его форм. Тройные точки (с жидкой фазой) лежат для Ga(I)-Ga(II) при 12 тыс. атм и 3 °С, а для Ga(II)-Ga(III) — при 30 тыс. атм и 45 °С.
Галлий весьма склонен к переохлаждению и его удалось удерживать в жидком состоянии до -40 °С. Многократное повторение быстрой кристаллизации переохлаждённого расплава может служить методом очистки галлия. В очень чистом состоянии (99,999 %) он был получен путём электролитического рафинирования, а также восстановлением водородом тщательно очищенного GaCl3. Высокая точка кипения и довольно равномерное расширение при нагревании делают галлий ценным материалом для заполнения высокотемпературных термометров. Несмотря на его внешнее сходство с ртутью, взаимная растворимость обоих металлов сравнительно невелика ( в интервале от 10 до 95 °С она изменяется от 2,4 до 6,1 атомного процента для Ga в Hg и от 1,3 до 3,8 атомного процента для Hg в Ga). В отличие от ртути жидкий галлий не растворяет щелочные металлы и хорошо смачивает многие неметаллические поверхности. В частности, это относится к стеклу, нанесением на которое галия могут быть получены зеркала, сильно отражающие свет (однако имеется указание на то, что очень чистый галий, не содержащий примеси индия, стекло не смачивает). Нанесением галия на пластмассовую основу используется иногда для быстрого получения радиосхем. Сплав 88 % Ga и 12 % Sn плавится при 15 °С, а некоторые другие содержащие галлий сплавы (например, 61,5 % Bi, 37,2 — Sn и 1,3 — Ga) были предложены для пломбирования зубов. Они не изменяют своего объёма с температурой и хорошо держатся. Галий можно использовать также как уплотнитель для вентелей в вакуумной технике. Однако следует иметь в виду, что при высоких температурах он агрессивен по отношению и к стеклу, и ко многим металлам.
Палочни металлического индия при сгибании хрустят подобно оловянным. На бумаге он оставляет тёмную черту. Важное применение индия связано с изготовлением германиевых выпрямителей переменного тока. Благодаря своей лёгкоплавкости он может играть роль смазки в подшипниках.
Введение небольшого количества индия в сплавы меди сильно повышают устойчивость к действию морской воды, а присадка индия к серебру усиливает его блеск и предупреждает потускнение на воздухе. Сплавам для пломбирования добавка индия придаёт повышенную прочность. Электрическое покрытие индием других металлов хорошо предохраняет их от коррозии. Сплав индия с оловом (1:1 по массе) хорошо спаивает стекло со стеклом или металлом, а сплав состава 24 % In и 76 % Ga плавится при 16 °С. Плавящийся при 47 °С сплав 18,1 % In с 41,0 % Bi, 22,1 % Pb, 10,6 % Sn и 8,2 % Cd находит медицинское использование при сложных переломах костей (вместо гипса).
Таллий используется главным образом для изготовления сплавов с оловом и свинцом, обладающих высокой кислотоупорностью. В частности, сплав состава70 % Pb, 20 % Sn и 10 % Tl хорошо выдерживает действие смесей серной, соляной и азотной кислот.
В сухом воздухе галлий близок и индий не изменяются, а талий покрывается серой плёнкой оксида. При прокаливании все три элемента энергично соединяются с кислородом и серой. С хлором и бромом они взаимодействуют кже при обычной температуре, с иодом — лишь при нагревании. Располагаясь в ряду напряжений около железа, Ga, In и Tl растворимы в кислотах.
По отношению к воде галлий и компактный индий устойчивы, а талий в присутствии воздуха сильно разрушается водой с поверхности. С азотной кислотой галлий реагирует лишь медленно, а таллий весьма энергично. Напротив серная кислота, и особенно соляная легко растворяет Ga и In, тогда как Tl взаимодействует с ними значительно медленнее (вследствие образования на поверхности защитной плёнки труднорастворимых солей). Растворы сильных щелочей легко растворяют галлий, лишь медленно действуют на индий и не реагируют с таллием. Галлий медленно растворяется также в NH4OH. Летучие соединения всех трёх элементов окрашивают бесцветное пламя в характерные цвета: Ga — в почти незаметный для глаза тёмно-фиолетовый (l= 417,1 нм), In — в тёмно-синий (l = 451,1 нм), Tl — в изумрудно-зелёный (l = 535,1 пм).
Галлий и индий, по-видимому, не ядовиты. Напротив, таллий сильно ядовит, причём по характеру действия похож на Pb и As. Поражает он нервную систему, пищеварительный тракт и почки. Симптомы строго отравления проявляются не сразу, а через 12-20 часов. При медленно развивающемся храническом отравлении (в том числе и через кожу) наблюдается прежде всего возбуждение и расстройство сна. В медицине препаратами таллия пользуются для удаления волос (при лишаях и т. п.). Соли таллия нашли применение в светящихся составах как вещества, увеличивающие продолжительность свечения. Они оказались также хорошим средством против мышей и крыс.
Обычная валентность галлия и индия равна трём. Таллий даёт производные, в которых от трёх- и одновалентен.
В ряду напряжений галлий располагается между Zn и Fe, а индий и таллий — между Fe и Sn. Переходом Ga и In по схеме: Э3+ + 3 е- = Э отвечают нормальные потенциалы: -0,56 и -0,33 В (в кислой среде) или -1,2 и 1,0 В (в щелочной среде). Таллий переводится кислотами в одновалентное состояние (нормальный потенциал -0,34 В). Переход Tl3+ + 2 e- = Tl+ характеризуется нормальным потенциалом +1,28 В в кислой среде или + 0,02 В — в щелочной.
Оксид галлия и его аналогов — белый Ga2O3, жёлтый In2O3 и коричневый Tl2O3 в воде нерастворимы. Отвечающие им гидроксиды — Э(OH)3 (которые могут быть получены исходя их солей) представляют собой студенистые осадки, практически нерастворимые в воде, но растворяющиеся в кислотах. Белые гидроксиды Ga и In растворимы также в растворах сильных щелочей с образованием аналогичных алюминатам галлатов и интатов. Они имеют, следовательно, амфотерный характер, причём кислотные свойства выражены у In(OH)3 слабее, а у Ga(OH)3 сильнее, чем у Al(OH)3. Так, помимо сильных щелочей, Ga(OH)3 растворим в крепких растворах NH4OH. Напротив, красно-коричневый Tl(OH)3 в щелочах не растворяется.
Теплоты образования оксидов Э2О3 галия и его аналогов уменьшается в ряду 1088 (Ga), 924 (In) и 389 (Tl). При нагревании на воздухе галлий практически окисляется только до GaО. Поэтому Ga2О3 обычно получают обезвоживанием Ga(ОН)3. Индий при нагревании на воздухе образует In2О3, а таллий — смесь Tl2О3 и Tl2О с тем большим содержанием высшего оксида, чем ниже температура. Нацело до Tl2О3 таллий может быть окислен действием озона.
Растворимость оксидов Э2О3 в кислотах увеличивается в ряду Ga-In-Tl. В том же ряду уменьшается прочность связи элемента с кислородом: Ga2О3 плавится при 1795 °С без разложения, In2О3 переходит в In3О4 лишь выше 850 °С, а мелко раздробленный Tl2О3 начинает отщеплять кислород уже около 90 °С. Однако для полного перевода Tl2О3 в Tl2О необходимы гораздо более высокие температуры. Под избыточным давлением кислорода In2О3 плавится при 1910 °С, а Tl2О3 — при 716 °С.
Теплоты гидротации оксидов по схеме:
Э2О3 + 3 Н2О = 2 Э(ОН)3
составляет + 92 кДж (Ga), + 4 (In) и - 188 (Tl). В соответствии с этим лёгкость отщепления гидроксидами воды возрастает от Ga до Tl: если Ga(ОН)3 полностью обезвоживается лишь при прокаливании, то Tl(ОН)3 переходит в Tl2О3 даже при стоянии под жидкостью, из которой он был выделен.
При нейтрализации кислотных растворов солей галлия его гидроксид осаждается приблизительно в интервале рН = 3-4. Свежелсаждённый Ga(OH)3, хорошо растворим в крепких растворах аммиака, но по мере старения растворимость всё более снижается. Её изоэлектрическая точка лежит при рН = 6,8, а ПР = 2·10-37. Для In(OH)3 было найдено ПР = 1·10-37, а для Tl(OH)3 — 2·10-45.
Для вторых и третьих констант диссоциации Ga(OH)3 по кислотному и основному типам были найдены следующие значения:
H3GaO3 К2 = 5·10-11 К3 = 2·10-12
Ga(OH)3 К2 = 2·10-11 К3 = 4·10-12
Таким образом, гидроксид галлия представляет собой случай электролита, очень близкого к идеальной амфотерности.
Различие кислотных свойств гидроксидов галия и его аналогов отчётливо проявляется при их взаимодействии с растворами сильных щелочей (NaOH, KOH). Гидроксид талий легко растворяется с образовагием галлатов типа M[Ga(OH)4], устойчивых и в растворе, и в твёрдом состоянии. При нагревании они легко теряют воду (соль Na — при 120, соль K — 137 °С) и переходят в состветствующие безводные соли типа MGaO2. Для получаемых из растворов галлатов двухвалентных металлов (Ca, Sr) характерен другой тип — M3[Ga(OH)6]2.
Гидроксид индия почти нерастворим в лисьных щелочах (NaOH, KOH), но при очень высоких их концентрациях образуются индаты типа M3[In(OH)6]2·2H2O, которые тоже почти нерастворимы. Водой они полностью гидролизуются.
Гидроксид таллия легко пептизуется сильными щелочами (с образованием отризательного золя), но нерастворима в них и таллатов не даёт. Сухим путём (сплавлением оксидов с соответствующими карбонатами) производные типа МЭО2 были получены для всех трёх элементов подгруппы галлия. Однако в случае таллия они оказались смесями оксидов.
Ионы Ga••• и In••• бесцветны, ион Tl••• имеет желтоватую окраску. Производящиеся от них соли большинства кислот хорошо растворимы в воде, но сильно гидролизованы. Из растворимых солей слабых кислот многие подвергаются практически полному гидролизу.
Эффективные радиусы ионов Ga3+, In3+ и Tl3+ равны соответственно 62, 92 и 105 пм. В водной среде они непосредственно окружены, по-видимому, шестью молекулами воды. Такие гидратированные ионы несколько диссоциированы по схеме Э(ОН2)6Ы Э(ОН2)5ОН•• + Н•, причём их константы диссоциации оцениваются в 3·10-3 (Ga) и 2·10-4 ( In).
Галогениды Ga3+, In3+ и Tl3+ в общем похожи на соответствующие соли Al3+. Кроме фторидов, они сравнительно легкоплавки и хорошо растворимы не только в воде, но и в ряде органических растворителей. Окрашены из них лишь жёлтые GaI3,
23-56
В то время, как производные низших валентнослей Ga и In для них не типичны, для галлия наиболее характерны именно те соединения, в которых он одновалентен. Поэтому соли Tl3+ имеют заметно выраженные окислительные свойства.
Гемиоксид талия Tl2O образуется в результате взаимодействия элементов при высоких температурах. Она представляет собой чёрный гигроскопичный порошок. С водой гемиоксид таллия образует жёлтый гигроскопичный порошок гидроксида таллия TlOH, который при нагревании легко отщепляет воду и переходит обратно в Tl2O.
Гидроксид таллия (I) хорошо растворим в воде и является сильным основаниям. Образуемые им соли в большинстве бесцветны и кристаллизуются без воды. Хлорид, бромид и иодид почти нерастворимы, но некоторые другие соли растворимы в воде. Производные TlOH и слабых кислот вследствие гидролиза дают в растворе щелочную реакцию. При действии сильных окислителей (например, хлорной воды) одновалентный таллий окисляется до трёхвалентного.
57-66
По химическим свойствам элементов и соединений подгруппа галлия во многом похожа на подгруппу германия. Так, для Ge и Ga более устойчива высшая валентность, для Pb и Tl низшая, химический характер гидроксидов в рядах Ge-Sn-Pb и Ge-In-Tl изменяются даже более тонкие черты сходства, например малая растворимость (Cl, Br, I) солей PbII, так и TlI. При всём том между элементами обеих подгрупп имеются и существенные (частично обусловленные их разной валентностью): кислотный характер гидроксида Ga и его аналогов выражен слабее, в противоположность PbF2 фтористый таллий хорошо растворим и т. д.
22
ПОДГРУППА ГЕРМАНИЯ.
Содержание элементов этой подгруппы в земной коре по ряду германий (2·10-4) — олово (6·10-4) — свинец (1·10-4%) изменяется лишь незначительно.
Германий принадлежит к весьма рассеянным элементам, и образование рудных скоплений для него не характерно. Богатые германием минералы — германит (Cu2S·CuS·GeS2) и аргиродит (4Ag2S·GeS2) — встречается редко. Следы германия были обнаружены во всех исследованных силикатах. Значительно большие количества этого элемента (до 1%) содержится иногда в золе каменных и бурых углей.
Основной формой природного олова является минерал касситерит(SnO2) или оловянный камень. Разработка оловянных руд рентабельна (т.е. экономически выгодна) уже при содержании в них 0,2 вес. % Sn. Важнейшей рудой свинца является галенит(PbS), иначе свинцовый блеск. Меньшее значение имеет минерал церуссит— PbCO3.
Германий был предсказан Д. И. Менделеевым в 1871 г., а открыт в 1886 г. Олово и свинец принадлежат к наиболее давно известным человечеству элементам: египтяне умели выплавлять их из руд более чем за 3000 лет до н. э. В Индии свинец стал известен около 2500 лет, а олово 1500 лет до н. э. Выплавка олова производилась и в древнем Китае.
Изотопный состав: германий — 70 (20,5%), 72 (27,4), 73 (7,8), 74 (36,5), 76 (7,8); олово — 112 (0,9%), 114 (0,7), 115 (0,3), 116 (14,2), 117 (7,6), 118 (24,0), 119 (8,6), 120 (33,0), 122 (4,7), 124 (6,0); обыкновенный свинец — 202 (следы), 204 (1,4%), 206 (25,2), 207 (21,7), 208 (51,7).
Среднее содержание элементов подгруппы германия в живых организмов невелико — порядка 10-6 вес. %. Однако некоторыми растениями свинец концентрируется настолько, что содержание его может доходить до 3 вес. %. Биологическая роль всех трёх элементов неизвестна, но имеется указание на то, что германий стимулирует деятельность костного мозга и селезёнки. Человеческий организм содержит около 2·10-5 олова и 1·10-4 вес. % свинца. Из отдельных частей тела наибольшее содержание Sn — в языке, а Pb — в длинных костях. Средний суточный рацион человека включает в себя около 17 мг Sn и 0,3 мг Pb. Оба элемента выводятся из организма главным образом с калом.
Добыча германия в большом масштабе ещё не производится. Получают его главным образом как побочный продукт при переработке некоторых цинковых руд. Выплавка олова ведётся путём восстановления касситерит углём. Галенит путём прокаливания на воздухе переводят в PbO, после чего полученный оксид восстанавливается до металла.
Из содержащих германий природных материалов выделяют в конечном счёте GeO2, который затем при температурах около 1000 °С восстанавливают водородом до металла. Простейшая схема промышленного восстановления свинца основывается на двух последовательных реакциях:
2 PbS + 3 O2 = 2 SO2 + 2 PbO + 844 кДж и затем
2 PbO + PbS + 234 кДж = SO2 + 3 Pb.
Очистка свинца может быть осуществлена путём электролиза. Электролитом служит раствор PbSiF6, в качестве анода берётся пластина технического металла, а на катоде осаждается чистый свинец (99,99%).
По физическим свойствам олово и свинец являются типичными металлами, а германий похож скорее на кремний. Некоторые их константы сопоставлены ниже.
| Ge | Sn | Pb | |
| Цвет | серовато- белый | серебристо- белый | голубоватый |
Плотность, г/см3 | 5,3 | 7,3 | 11,3 |
Температура плавления °С | 937 | 232 | 328 |
Температура кипения °С | 2850 | 2720 | 1751 |
| Электропроводность (Hg = 1) | 0,001 | 8 | 5 |
Твёрдость и хрупкость рассматриваемых элементов быстро уменьшается по ряду Ge–Sn–Pb: германий очень тверд и хрупок, свинец царапается ногтём и прокатывается в тонкие листы. Олово занимает промежуточное положение. Все элементы подгруппы германия легко дают сплавы между собой и со многими другими металлами. В некоторых случаях при сплавлении образуются химические соединения (например, типа Mg2Э).
Пары олова и свинца состоят почти исключительно из одноатомных молекул, а у германия (при температурах 1600ч2000 К) содержат также полимеры Gen, где n = 2ч7. Энергия связи GeGe в Ge2 cоставляет 272 кДж/моль.
Элементарный германий имеет структуру алмаза. Под высокими давлениями германий может существовать в трёх других аллотропных формах. Все они имеют различные кристаллические структуры, повышенную плотность (до 6,0 г/см3) и значительно лучшую электропроводность. В обычных условиях эти модификации неустойчивы.
Выше 550 °С германий становится пластичным и поддается механической обработке. Плавление его сопровождается увеличением плотности (на 5%) и электропроводности (примерно в 15 раз). По мере повышения давления температура плавления германия последовательно снижается и при 180 тыс. атм становится равной 347 °С. Электросопротивление чистого германия с повышением давления возрастает (но при 115 тыс. атм. он приобретает свойства металла), а у олова и свинца оно уменьшается.
Для обычной формы олова характерна структура, в которой каждый его атом имеет четырёх соседей на расстояниях 302 пм и ещё двух на расстояниях 318 пм. Для свинца имеет место структура, в которой каждый его атом имеет 12 равноотстоящих на 350 пм соседей. В отличие от германия температуры плавления обоих металлов с повышением давления возрастают (у свинца при 30 тыс. атм. приблизительно до 520 °С).
Сгибание оловянных палочек сопровождается характерным хрустом, обусловленным трением отдельных кристаллов друг о друга. При нагревании Sn выше 160 °С происходит укрупнение этих кристаллов (без изменения структуры), сопровождающееся резким ослаблением их сцепления друг с другом. В результате плотность металла падает (от 7,3 до 6,6 г/см3), он становится очень хрупким и его можно легко растереть в мелкий порошок.
Кроме обычного олова известна устойчивая ниже +13 °С его аллотропическая форма имеющая структуру алмаза [d(SnSn) = 281 пм] и представляющая собой серый порошок с плотностью 5,8 г/см3. Теплота перехода в неё обычного олова составляет лишь 2 кДж/моль, а скорость перехода ничтожно мала. Такой переход, сопровождающийся превращением оловянного предмета в серый порошок, при охлаждении олова обычно не происходит. Однако он наблюдается на некоторых старинных сосудах и медалях из олова.
Скорость перехода в серую модификацию несколько зависит от природы примесей (например, цинк её увеличивает, а свинец уменьшает) и сильно повышается с понижением температуры, достигая максимума при –33 °С. При нахождении Sn в растворе его соли такой переход довольно быстро происходит уже около 0 °С. Превращение гораздо легче наступает при соприкосновении обычного олова с уже превращённым. Поэтому возможно “заражение” оловянных предметов друг от друга и распространение таким образом “болезни”, очень метко названной “оловянной чумой”. Последняя нередко наблюдалась в средние века, когда домашняя посуда зажиточных слоёв населения изготовлялась преимущественно из различных сплавов на основе олова. Чаще всего страдали также делавшиеся из довольно чистого олова органные трубы. Из-за разрушения паянных оловом сосудов с жидким топливом в 1912 г. погибла экспедиция Скотта к Южному полюсу. С оловянной чумой приходится особенно считаться при хранении запасов олова.
Кристаллы серого олова могут быть получены из насыщенного раствора в ртути при –65 °С. Они обладают полупроводниковыми свойствами и характеризуются особой чувствительностью к инфракрасным лучам (до 15 мк). Добавкой 0,75% германия область практической устойчивости серого олова может быть повышена до +60 °С.
Все три элемента весьма важны для современной техники. Значительное применение находят также некоторые соединения олова и свинца. Производные свинца сильно ядовиты.
Германий является типичным полупроводником (n-типа с шириной запрещённой зоны 0,75 эВ) и находит разнообразное использование в электротехнике. Наиболее широко он применяется для изготовления выпрямителей переменного тока. Применение это основано на униполярной проводимости, возникающей при контакте между чистым германием и сплавом германия с индием. Ток (поток электронов) проходит в такой установке практически только от германия к сплаву, но не наоборот. Германиевые выпрямители характеризуются чрезвычайно высоким (порядка 98%) коэффициентом полезного действия и очень большим (при правильной эксплуатации) сроком службы. Основным недостатком таких выпрямителей является их чувствительность к нагреванию — выше 70 °С их эффективность быстро падает.
Важной областью использования германия является инфракрасная оптика, так как лучи с длиной волны больше 2 мк он практически не задерживает. Напротив, в световом и близких к нему диапазонах (0,2 ч 2 мк) германий интенсивно поглощает энергию. Если блестящую металлическую поверхность (которая хорошо хранит тепло, но плохо нагревается) покрыть пленкой германия, то поверхность нагревается гораздо сильнее, чем без плёнки. Сообщалось, что в подготовленной таким образом бочке под действием солнечного света можно получить кипяток.
Для применения германия в качестве полупроводника он должен быть очень чист. Например, содержание As не может превышать 10-7%, т.е. одного атома As на миллиард атомов германия. Достижение столь высокой чистоты требует прежде всего тщательной очистки материала, из которого вырабатывается вещество полупроводника. Однако большей частью дополнительной очистке приходится подвергать и само это вещество.
При частичной кристаллизации расплавленного вещества примеси неодинаково распределяются между твёрдой и жидкой фазами. Чаще они концентрируются в расплаве. Если находящийся в тугоплавкой лодочке расплав последовательно охлаждать с одной стороны, то примеси оттесняются к концу, затвердевающему последним. Удалив затем этот конец слитка, получают вещество более чистое, чем оно было первоначально. При другом, более совершенном варианте — зонной плавке — нагреву до плавления последовательно подвергаются отдельные участки помещённого в тугоплавкой лодочке вещества, и перемещающаяся зона расплава несёт с собой примеси в один конец слитка. Перед простой направленной кристаллизацией зонная плавка имеет то большое преимущество, что с одним и тем же слитком может быть повторяема многократно, причём процесс этот легко поддаётся автоматизации. Во избежание окисления очищаемого вещества кристаллизацию проводят в вакууме или инертной атмосфере.
На том же принципе основан один из способов выращивания монокристалла. Для этого на поверхность расплава, нагретого чуть выше температуры плавления, помещают кристалл данного вещества, который затем медленно поднимают автоматическим устройством. Таким путём может быть не только очищено исходное вещество, но и получен его монокристалл значительной величины (например, были получены образцы Ge диаметром 5 см и длиной 18 см или Si диаметром 2 см и длиной 24 см).
С помощью рассмотренных методик германий удавалось доводить до чистоты в 10 девяток. Те же методики позволяют решать и очень важную обратную задачу — равномерно распределять в очищенном веществе нужное для успешной работы полупроводника заранее задаваемое количество определённых примесей.
Олово используется главным образом для лужения железа с целью предохранения его от ржавления (белая жесть для консервной промышленности). Толщина таких оловянных покрытий очень мала — порядка микронов. В виде тонких листков (т. н. станниоля) олово потребляется для изготовления конденсаторов в электротехнической промышленности. Свинец применяется для изготовления аккумуляторных пластин, обкладок электрических кабелей, пуль и дроби, для защиты от рентгеновского излучения и g-лучей, а также в химической промышленности (трубопроводы и т. д.). Очень большие количества олова и свинца расходуются на изготовление ряда технически важных сплавов.
Важнейшими из них являются различные бронзы (сплавы Cu и Sn), сплавы для подшипников (баббиты, изготовляемые обычно на основе Pb или Sn и содержащие так же Sb и Cu), типографские сплавы (5—30% Sn, 10—20% Sb, остальное Pb) и обычный “мягкий” припой (30—70% Sn, 70—30% Pb). Его заменителем часто может служить более дешевый сплав состава 90% Pb, 6% Sn, 4% Sb. Большое значение имеют сплавы для подшипников приблизительного состава 98% Pb, 1% Ca, 1% Na.
Ежегодная мировая добыча германия составляет около 100 т. Мировая добыча олова и свинца составляла соответственно (в тысячах тонн): 4 и 30 в 1800 г., 85 и 875 в 1900 г., 166 и 1750 в 1950 г. С тех пор она стабилизировалась примерно на том же уровне.
Летучие соединения свинца окрашивают бесцветное пламя газовой горелки в бледно-синий цвет. Будучи постоянно вводим в организм даже очень небольшими дозами, он накапливается (частично замещая кальций костного скелета), причём ядовитое действие его постоянно усиливается. Свинцовое отравление иногда фигурирует как профессиональная болезнь лиц, постоянно имеющих дело со сплавами или препаратами свинца (например, типографских наборщиков). Первыми симптомами хронического отравления являются образование серой каймы на дёснах и боли в области живота. В дальнейшем развиваются различные расстройства нервной системы. Максимально допустимое содержание Рb в воздухе производственных помещений составляет 10-5 мг/л. Острое отравление свинцовыми препаратами вызывает тяжёлые поражения пищеварительного тракта. В качестве средства первой помощи при остром отравлении применяют разбавленный раствор H2SO4.
Под действием кислорода воздуха германий и олово не изменяются, а свинец окисляется. Поэтому свинцовые предметы всегда покрыты синевато-серым слоем оксида и не имеют блестящего металлического вида. Плёнка оксида в обычных условиях хорошо предохраняет металл от дальнейшего окисления, но при нагревании оно идёт дальше, и свинец постепенно окисляется нацело. При нагревании на воздухе начинает окисляться и олово. Германий взаимодействует с кислородом лишь выше 700 °С. Все три элемента способны соединяться с галогенами и серой. Вода не действует на германий и олово. Со свинца она постепенно снимает оксидную плёнку и тем самым способствует его дальнейшему окислению. Лучшим растворителем свинца является разбавленная азотная кислота, германия и олова — царская водка. Взаимодействие с ней обоих элементов идёт по схеме:
2 Э + 4 HNO3 + 12 HCl = 3 ЭCl4 + 4 NO + 8 H2O.
В ряду напряжений Ge располагается между медью и серебром, а Sn и Pb — непосредственно перед водородом. Поэтому они вытесняются из солей многими металлами (например, цинком).
Отношение элементов подгруппы германия к отдельным кислотам существенно различается. Соляная кислота не действует на германий. Олово лишь очень медленно растворяется в разбавленной НCI, тогда как с концентрированной легко (особенно при нагревании) идёт реакция по схеме:
Sn + 2 HCl = SnCl2 + H2.
Свинец при взаимодействии с НСI покрывается слоем труднорастворимого РbCI2, препятствующим дальнейшему растворению металла. Аналогично идёт взаимодействие и с серной кислотой до тех пор, пока крепость её не превышает 80%. При более высоких концентрациях Н2SO4 образуется растворимая кислая соль Pb(HSO4)2 (или комплексная кислота H2[Pb(SO4)2]), уже не защищающая свинец от дальнейшего действия серной кислоты. На германий разбавленная серная кислота не действует, на Sn — почти не действует. В горячей концентрированной H2SO4 оба элемента растворяются по схемам:
Э + 4 H2SO4 = Э(SO4)2 + 2 SO2 + 4 H2O.
При действии на германий азотной кислоты образуется осадок гидрата диоксида — хGeO2·уH2O. Аналогично — по схеме:
Sn + 4 HNO3 = SnO2 + 4 NO2 + 2 H2O
— действует концентрированная кислота и на олово. Напротив, в сильноразбавленной холодной азотной кислоте олово медленно растворяется с образованием Sn(NO3)2. Водород при этом не выделяется, а идёт на восстановление азотной кислоты. При действии НNO3 на свинец по реакции:
3 Pb + 8 HNO3 = 3 Pb(NO3)2 + 2 NO + 4 H2O
образуется Pb(NO3)2. Соль эта нерастворима в концентрированной HNO3 и предохраняет металл от дальнейшего действия кислоты. Напротив, в воде она хорошо растворима, и поэтому в разбавленной азотной кислоте свинец растворяется.
Растворы щелочей на германий почти не действуют (но при одновременном наличии Н2О2 он легко растворяется). При отсутствии окислителей олово и свинец медленно растворяются в сильных щелочах по схеме:
Э + 2 NaOH = Na2ЭО2 + Н2.
Растворимостью олова в щелочах пользуются для снятия его со старых консервных банок, после чего металл выделяют из раствора электролитически. Практически такое растворение (обычно при добавке метанитробензойной кислоты) осуществляется по схеме:
Sn + 2 NaOH + O2 = Na2SnO3 + H2O.
Ввиду высокой стоимости олова его регенерация (обратное получение) имеет большое экономическое значение.
На устойчивость свинца по отношению к воде сильно влияет содержание в последней растворенного углекислого газа. Небольшие его концентрации способствуют устойчивости свинца из-за образования на его поверхности слоя практически нерастворимого PbCO3. Напротив, при более высоких концентрациях СО2 образуется кислый углекислый свинец Pb(HCO3)2, переходящий в раствор. Использование содержащей его воды для питья ведёт к постепенному развитию свинцового отравления. В древнем Риме, где для водопроводов применялись свинцовые трубы, такое отравление было, по-видимому, весьма распространённым. На это указывают результаты анализа останков древних римлян.
Характерные для германия и его аналогов валентности — 4 и 2. Для германия более типичны те соединения, в которых он четырёхвалентен. При обычных условиях производные четырёхвалентного Sn более устойчивы. Напротив, для свинца значительно более типичны соединения, в которых он двухвалентен.
В связи с этим производные двухвалентных Ge и Sn являются восстановителями (притом очень сильными), а соединения четырёхвалентного Pb — окислителями (также очень сильными). Но переход от более низкой к более высокой положительной валентности, как правило, легче идёт в щелочной среде, а обратный переход — в кислой. Поэтому восстановительные свойства двухвалентных Ge и Sn в щелочной среде выражены сильнее, чем в кислой, а четырёхвалентный Pb, будучи очень сильным окислителем в кислой среде, в щелочной таковым не является.
Для элементов подгруппы германия известны оксиды типов ЭО и ЭО2. При прокаливании на воздухе Ge и Sn образуют их высшие оксиды, а при прокаливании свинца получается низший. Остальные оксиды получают лишь косвенным путём.
Все рассматриваемые оксиды представляют собой твёрдые вещества. Монооксиды германия и олова характеризуются чёрной окраской, PbO — жёлтовато-красной, GeO2 и SnO2 — белой, PbO2 — темно-коричневой. В воде они почти нерастворимы.
Монооксид германия может быть получен по протекающей при 700ё900 °С реакции:
СО2 + Ge = GeO + CO.
При этих температурах он летуч и осаждается на охлаждаемой поверхности в виде аморфного светло-жёлтого порошка. Диоксид германия (т. пл. 1116, т. кип. 1200 °С) является обычным исходным веществом при получении металлического германия. Напротив, SnO2 (т. пл. 1630 °С) и PbO (т. пл. 886, т. кип. 1580 °С) готовят прокаливанием металлов на воздухе. Монооксид олова получают нагреванием раствора SnCl2 cо щёлочью. В твёрдом состоянии он имеет тенденцию к дисмутации по схеме:
2 SnO = SnO2 + Sn,
но в жидком (т. пл. 1040 °С) и газообразном (т. кип. 1425 °С) устойчив. Помимо обычной чёрной известны метастабильные синяя и красная формы SnO. Для получения PbO2 обычно применяется взаимодействие уксуснокислого свинца с белильной известью, протекающее по схеме:
Рb(CH3COO)2 + Ca(Cl)OCl + H2O = PbO2Ї + CaCl2 + 2 CH3COOH.
При нагревании РbO2 происходит последовательное образование низших оксидов свинца:
PbO2 (290-320 °С) ® Pb2O3 (390-420 °С) ® Pb3O4 (530-550 °С) ® РbO.
Диоксид германия имеет большое значение для промышленности оптического стекла, так как при частичной замене им диоксида кремния получаются очень прозрачные и сильно преломляющие свет стёкла. Диоксид олова используется в керамической промышленности при изготовлении эмалей и глазурей, а также употребляется для получения стекла. Стекло с поверхностным слоем из SnO2 обладает полупроводниковой проводимостью. Диоксид свинца (иногда неправильно называемый пероксидом) употребляется в спичечной промышленности. Диоксид олова применяется в стекольном производстве (для получения рубинового стекла) и при ситцепечатании (как восстановитель). Монооксид свинца находит медицинское использование (свинцовый пластырь) и потребляется рядом отраслей промышленности, а также для изготовления в смеси с глицерином замазки для металла, стекла и камня.
Свинцово-глицериновая замазка готовится тщательным смешиванием хорошо высушенного при 300 °С свинцового глёта с безводным глицерином (в весовом соотношении 5:1). Она схватывается через 30-40 минут и через несколько часов твердеет (вследствие образования глицератов свинца). Получающаяся твёрдая масса газо- и водонепроницаема, обладает механической прочностью и выдерживает нагревание почти до 300 °С. Подлежащие соединению поверхности следует перед нанесением замазки протереть глицерином.
Так как с водой эти оксиды почти не соединяются, отвечающие им гидроксиды получают обычно действием сильных щелочей на растворы соответствующих солей, например, по реакциям
SnCl4 + 4 NaOH = 4 NaCl + Sn(OH)4
Pb(NO3)2 + 2 NaOH = 2 NaNO3 + Pb(OH)2.
Они выделяются в виде аморфных осадков белого цвета (кроме бурого Pb(OH)4). В воде Ge(OH)4 заметно растворим, тогда как растворимость остальных очень мала.
По химическим свойствам все эти гидроксиды представляют собой амфотерные соединения. Диссоциация их растворённой части протекает в конечном счёте (если не считаться с её постепенностью) по схемам
Э••+ 2 ОН’ Ы Э(ОН)2є Н2ЭО2Ы 2 Н• + ЭО2”
Э•••• + 4 ОН’Ы Э(ОН)4є Н4ЭО4Ы 2 Н• + ЭО3” + Н2О.
Относительная характерность того или иного направления диссоциации отдельных представителей видна из следующего приблизительного сопоставления:
ЬУвеличение кислотных свойств
Ge(OH)4 Sn(OH)4 Pb(OH)4
Ge(OH)2 Sn(OH)2 Pb(OH)2
усиление основных свойствЮ
Наиболее отчётливо кислотные свойства выражены у гидроксида германия (IV), который всё же является очень слабой кислотой. Основные свойства наиболее отчётливо выражены у Pb(OH)2, который сообщает воде заметную щелочную реакцию.
Ввиду своего амфотерного характера рассматриваемые гидроксиды способны растворятся и в сильных щелочах, и в кислотах. При действии на них щелочей образуются соли типа М2ЭО3 или М2ЭО2, содержащие Ge, Sn или Pb в составе аниона, а при действии кислот — соли этих элементов с катионами Э2+ или Э4+.
Гидратные формы Э(ОН)2 и Э(ОН)4 являются простейшими. В действительности осадки гидроксидов содержат переменные количества воды, и их состав выражается более общими формулами хЭО·уН2О и хЭО2·уН2О. Для некоторых гидратных форм известны отвечающие им комплексные соединения. Например, для SnO2·4H2O получены cоли комплексной молибдо-оловянной кислоты типа М8[Sn(Mo2O7)6], где М — одновалентный металл. Аналогичная гетерополикислота известна и для германия.
В процессе постепенной нейтрализации разбавленных (0,01-0,1 М) кислых растворов солей двухвалентных олова и свинца Sn(OH)2 (ПР = 1·10-26) и Pb(OH)2 (ПР = 1·10-15) начинают осаждаться соответственно при рН 2 и 6. Константа первой ступени основной диссоциации Pb(OH)2 равна 1·10-3, а кислотной — 1·10-11, т.е. на каждую диссоциированную по кислотному типу молекулу приходится 100 млн. молекул, диссоциированных по основному типу. Константы второй ступени основной диссоциации (ЭОН•Ы Э•• + ОН’) для Sn(OH)2 и Pb(OH)2 равны соответственно 1·10-12 и 2·10-8. Производящиеся от диоксида германия кислоты имеют две формы — Н2GeO3 (К1 = 1·10-9, К2 = 2·10-13) и H2Ge5O11 (К1 = 6·10-7, К2 = 2·10-8), однако существование второй из них не бесспорно.
Гидрат диоксида олова имеет характер геля. Свежеосаждённый (например, действием NaOH на SnCI4) он содержит много воды и при исследовании рентгеновскими лучами не показывает кристаллической структуры. При стоянии над раствором или нагревании происходит его постепенное старение. Процесс заключается в полимеризации молекул хSnO2·yH2O, идущий с отщеплением воды. В результате получаются всё более крупные и бедные водой частицы. На известной стадии старения анализ при помощи рентгеновских лучей уже обнаруживает в геле микроструктуру (отвечающую структуре SnO2). Подобные гели с ясно выраженной внутренней кристаллической структурой могут быть получены и непосредственно — они образуются при действии концентрированной HNO3 на металлическое олово.
По мере старения геля SnO2 идёт изменение не только его физических, но и химических свойств. Различие последних для крайних случаев — свежеосаждённого геля и сильно состарившегося — столь велико, что их приходится рассматривать в отдельности. Свежеосаждённую из солей форму называют обычно a-оловянной кислотой, а сильно состарившуюся (или полученную действием концентрированной HNO3 на олово) — b-оловянной. Тогда как переход a-формы в b-форму постепенно идёт самопроизвольно, обратный переход может быть осуществлён лишь сплавлением b-формы со щёлочью и последующей обработкой сплава кислотой.
Отношение этих форм к HCl и KOH:
a-Оловянная кислота | b-Оловянная кислота |
При действии концентрированной HCl легко растворяется с образованием SnCl4 | Под действием конц. НCl заметного изменения с осадком не происходит. При последующем разбавлении водой осадок пептизуется и образуется прозрачный золь. Прибавление к последнему конц. HCl, сопровождается коагуляцией и обратным выпадением b-оловянной кислоты в осадок. |
При действии раствора КОН (как крепкого, так и разбавленного) легко растворяется с образованием К2SnO3. Соль эта может быть получена и в кристаллическом состоянии (К2SnO3·3H2O). | В крепком растворе КОН не растворяется. При последующем сильном разбавлении водой осадок пептизуется и образует прозрачный золь. Кристаллические соли из последнего получены быть не могут. Упаривание золя ведёт к образованию геля SnO2, cодержащего адсорбированную щёлочь. |
Гидроксид четырёхвалентного свинца настолько легко теряет воду, что практически нацело переходит в РbO2 уже при своём образовании.
Гидроксид двухвалентного германия может быть получен восстановлением фосфористой кислотой раствора GeO2 в крепкой HCl с последующим осаждением избытком аммиака. Все операции проводятся в атмосфере азота. Выделяется Ge(OH)2 в виде рыхлого осадка, цвет которого (белый, жёлтый или красный) зависит от условий получения. Растворимость этого гидроксида в HCl выше, чем в NaOH, т.е. основные её свойства преобладают над кислотными. С помощью инфракрасной спектроскопии было показано, что структура сухого гидроксида двухвалентного германия действительно отвечает формуле Ge(OH)2. При нагревании до 350 °С она переходит в коричнево-чёрный GeO.
От гидрата PbO2 как кислоты, и Pb(OH)2 как основания, производятся два смешанных оксида свинца — Pb2O3 оранжевого цвета и Pb3O4(сурик) ярко-красного цвета. Первый является свинцовой солью метасвинцовой кислоты (H2PbO3), а второй — ортосвинцовой кислоты (H4PbO4). Таким образом, оба оксида — PbPbO3 и Pb2PbO4 — одновременно содержат в своём составе атомы свинца различной валентности. В воде они практически не растворимы.
Структура обоих промежуточных оксидов свинца может быть обоснована результатами их взаимодействия с разбавленной азотной кислотой. Так, из сурика две трети всего свинца растворяются, переходя в Pb(NO3)2, тогда как остальная треть остаётся в виде PbO2. Этим доказывается наличие в молекуле сурика двух атомов двухвалентного свинца и одного атома четырёхвалентного. Аналогично обосновывается и структура плюмбита свинца.
Оба соединения могут быть получены смешиванием щелочных растворов Pb(OH)2 и Pb(OH)4. В присутствии небольших концентраций избыточной щелочи при этом выпадает плюмбит свинца (в виде гидрата Pb2O3·3H2O), а при её больших концентрациях — сурик. В технике последний получают нагреванием PbO на воздухе до 450-500 °С, причём происходит присоединение к PbO кислорода. Порошок сурика в смеси с льняным маслом иногда употребляется в качестве замазки для придания стыкам труб газо- и водонепроницаемости.
Соли кислот типа Н2ЭО3 носят названия соответственно германатов, станнатов и плюмбатов. Большинство их бесцветно и малорастворимо в воде. Немногие растворимые соли (Na, K и др.) в растворах сильно гидролизованы. Кристаллический станнат натрия (Na2SnO3·3Н2О) находит применение при крашении тканей.
Кроме солей мета-кислот, производных от гидратной формы Н2ЭО3 (т.е. ЭО2·Н2О), для рассматриваемых элементов известны так же соли, отвечающие орто-кислотам Н4ЭО4 (т.е. ЭО2·2Н2О) и комплексным гексагидроксо-кислотам Н2[Э(ОН)6] (т.е. ЭО2·4Н2О). К солям последнего типа принадлежит станнат натрия (Na2[Sn(OH)6]), равно как и многие другие станнаты. Этот тип соединений является основным (по крайней мере в растворе) также для плюмбатов и германатов. При нагревании до 100-150 °С гидроксосоли обезвоживаются по схеме:
Na2[Э(ОН)6] = 3 Н2О + Na2ЭО3.
Образующиеся безводные соли, весьма тугоплавки (например, Na2GeO3 плавится при 1083 °С). Термическое разложение станната калия (в токе сухого азота) протекает по схемам:
3 K2SnO3 = 2 K2O + K2Sn3O7 (выше 830 °С)
и затем K2Sn3O7 = K2O + 3 SnO2 (выше 900 °С).
Аналогичные соли — K2PbO3 и K2Pb3O7 — известны и для свинца. Так как термическая устойчивость многих плюмбатов гораздо выше, чем у PbO2, они могут быть получены накаливанием на воздухе смеси PbO с оксидом (или гидроксидом) соответствующего металла.
Крашение тканей из естественных волокон осуществляется либо непосредственно за счёт прочной адсорбции краски на их поверхности, либо путём отложения частиц краски внутри имеющихся в волокнах пор. Последнее достигается при помощи различных методов. В одних случаях ткань пропитывают коллоидным раствором краски (каковым и является водный раствор многих органических красителей) и затем действием электролитов вызывают коагуляцию этого раствора, в других — ткань пропитывают раствором того или иного вещества, которое само не является краской, но путём соответствующей химической обработки (например, действием окислителей) может быть затем переведено в нерастворимую краску, остающуюся заключённой внутри пор волокна. Тогда эти приёмы неприменимы, пользуются так называемым протравным крашением, при котором на ткани предварительно осаждают вещества, прочно удерживаемые волокнами, с одной стороны, и хорошо адсорбирующие краску — с другой. К подобным веществам относятся многие гидроксиды (в частности, хSnO2·yH2O); в качестве протрав применяют дающие их при гидролизе соли (например, Na2[Sn(OH)6]). Искусственные волокна могут быть окрашиваемы в жёлтый цвет уже при их получении.
Соли кислот типа Н2ЭО2 носят название соответственно германитов, станнитов и плюмбитов. По свойствам они в общем похожи на германаты, станнаты и плюмбаты, но значительно менее устойчивы и в растворах гидролизуются ещё сильнее. При действии окислителей они легко переходят в соли соответствующих кислот типа Н2ЭО3. Особенно это относится к германитам и станитам, которые являются очень сильными восстановителями. Напротив, гидроксид трёхвалентного висмута восстанавливается станитом до металла:
2 Вi(OH)3 + 3 Na2SnO2 = 3 Na2SnO3 + 2 Bi + 3 H2O.
Реакция эта находит использование в аналитической химии.
Для предупреждения гидролиза станнитов растворы их должны содержать избыток щелочи. Если концентрация последней невелика, в растворе медленно идёт реакция распада по схеме:
NaHSnO2 = NaOH + SnO.
В результате раствор станита при стоянии (быстрее — при нагревании) приобретает чёрную окраску. В присутствии большого избытка щёлочи реакция распада идёт по схеме:
2 NaHSnO2 = Na2SnO3 + Sn + H2O.
Вследствие выделения мелкораздробленного олова раствор при этом направлении процесса окрашивается в чёрный цвет. Аналогичная реакция характерна и для германитов, но в сильнощелочной среде преобладает их распад по схеме:
NaHGeO2 + NaOH = Na2GeO3 + H2
Основной формой существования германитов, станнитов и плюмбитов в растворах щелочей являются, вероятно, M[Sn(OH)3], где М –– одновалентный металл. Некоторые станниты этого типа –– Na[Sn(OH)3], Ba[Sn(OH)3]2 и др. — были выделены в кристаллическом состоянии. Вместе с тем сплавлением PbO с NaOH был получен плюмбит состава Na2PbO2 ( т. пл. 820 °С ).
Для солей типа МНЭО2 элементов четвертой группы, вообще говоря, возможна таутомерия по схеме:
Н-О-Э-О-М Ы О=Э(Н)-О-М.
В ряду элементов Pb-Sn-Ge-Si-C производным Pb (плюмбитам) отвечает первая из этих структур, производным С (солям муравьиной кислоты) –– вторая. Из промежуточных элементов для Si также характерна вторая структура, тогда как для Ge и Sn вероятно наличие равновесия обеих форм.
Некоторые физические свойства галогенидов ЭГ4 сопоставлены ниже:
GeF4 | GeCl4 | GeBr4 | GeI4 | SnF4 | SnCl4 | SnBr4 | SnI4 | PbF4 | PbCl4 | |
| Теплота образования кДж/моль | 1187 | 539 | 347 | 142 | 527 | 405 | 940 | 330 | ||
| Длина связи, пм | 167 | 211 | 229 | 250 | 228 | 244 | 264 | 243 | ||
Энергия связи Э-Г, кДж/моль | 335 | 284 | 209 | 343 | 272 | 196 | ||||
| Цвет | бесцв. | бесцв. | бесцв. | красн. | бесцв. | бесцв. | бесцв. | жёлт. | бесцв. | жёлт. |
Т плавл., °С | -15 давл. | -50 | +26 | 147 | -33 | +30 | 145 | 600 | -15 | |
Т кип., °С | -37 возг. | +83 | 187 | 377 разл. | 705 возг. | 113 | 205 | 344 |
В отличие от газообразного при обычных условиях GeF4, SnF4 и PbF4 представляют собой очень гигроскопичные кристаллические вещества. Молекулы остальных рассматриваемых соединений имеют строение правильных тетраэдров с показанными выше ядерными расстояниями.
Тетрафторид германия имеет резкий (типа чесночного) запах и дымит на воздухе. Для него известен бесцветный, очень гигроскопичный кристаллогидрат GeF4·3H2O. Тетрахлорид германия почти нерастворим в концентрированной НCl, но хорошо растворяется во многих органических растворителях (а также в жидком SO2). Водой он гидролизуется, а с сухим аммиаком реагирует по схеме:
GeCl4 + 6 NH3 = 4 NH4Cl + Ge(NH)2.
Красный цвет GeI4 при охлаждении до -10 °С изменяется на оранжевый, а при температуре жидкого воздуха — на бледно-жёлтый. Выше точки плавления начинается распад по схеме:
GeI4 = GeI2 + I2.
Акцепторные свойства тетрагалогенидов германия выражены сильнее, чем у соответствующих тетрагалогенидов кремния.
Взаимодействием паров GeCl4 с порошком металлического германия при 430 °С был получен бесцветный кристаллический Ge2Cl4 (т. пл. 41 °С). Его давление пара при обычных температурах составляет 3 мм рт. ст. Водой Ge2Cl6 разлагается на НСl и нелетучее белое вещество, имеющее состав (GeOOH)2 аналогичное силикощавелевой кислоте.
Наиболее практически важным из галогенидов ЭГ4 является четырёххлористое олово, которое было впервые описано Либавием. В технике его обычно получают обработкой использованных жестяных консервных банок сухим хлором. Последний не действует на железо, а покрывающее его тонким слоем олово легко образует SnCl4. Четырёххлористое олово дымит на воздухе (вследствие гидролиза за счёт содержащейся в атмосфере влаги). Оно легко смешивается со многими малополярными растворителями и само является хорошим растворителем для многих неэлектролитов (I2, P, S и др.). Из водного раствора четырёххлористое олово выделяется обычно (при температурах 19-56 °С) в виде бесцветного кристаллогидрата SnCl4·5H2O. Из различных продуктов присоединения к хлорному олову кристаллический SnCl4·2OРCl3 (т. пл. 59, т. кип. 117 °С) интересен тем, что дополняющие координацию Sn до октаэдра атомы кислорода находятся в цис-положении друг к другу, т.е. обе молекулы хлороксида фосфора располагаются рядом.
Фторид четырёхвалентного свинца может быть получен действием фтора на PbF2 при 250 °С. Он крайне чувствителен к влаге и на воздухе тотчас буреет (переходя в PbO2). Четырёххлористый свинец образуется в результате взаимодействия PbO2 и крепкой HCl при охлаждении. Он очень неустойчив и распадается на РbCl2 и Cl2 под действием света и в присутствии даже следов влаги. Бромид и иодид четырёхвалентного свинца не получены.
Из производных рассматриваемых элементов, содержащих одновременно кислород и галоген, интересен аналогичный по составу фосгену оксохлорид германия GeOCl2. Это бесцветная маслянистая жидкость, нерастворимая в обычных растворителях (т. пл. -56 °С). Водой GeOCl2 быстро разлагается с образованием Ge(OH)4. Продуктами термического разложения GeOCl2 являются хлор и GeO. Последний получается в виде жёлтой модификации, которая выше 650 °С переходит в обычную чёрную.
Существование GeOCl2 было поставлено под сомнение. При этом имеется указание на возможность образования GeOF2 при взаимодействии GeF4 c SO2, а по схеме:
ЭСl4 + Cl2O = 2 Cl2 + ЭОСl2
были получены оксохлориды олова и свинца. Лучше изученный SnOCl2 представляет собой белый, весьма гигроскопичный порошок, при 155 °С разлагающийся на SnO2 и SnCl4. Он тримерен и имеет циклическое строение.
Цианид четырёхвалентного германия был получен по схеме:
GeI4 + 4 AgCN = 4 AgI + Ge(CN)4.
Он представляет собой белое твёрдое вещество, при взаимодействии с водой или нагревании выше 80 °С разлагающееся. В растворе КСN могут образовываться ионы [Ge(CN)6]”.
Самым характерным свойством галогенидов ЭГ4 является их сильно выраженная склонность к реакциям присоединения. Так, SnCl4 легко образует комплексы с НСl, H2O, NH3, оксидами азота, PCl3 и т. д., равно как со спиртами, эфирами и многими другими органическими соединениями. Весьма устойчивы соли комплексных кислот типа Н2SnГ6. Например, из смеси растворов SnCl4 и NH4Cl кристаллизуется соль состава (NH4)2[SnCl6], раствор которой показывает нейтральную реакцию на лакмус. Будучи взята в достаточно высоких концентрациях, Н2SnCl6 заметно не разлагается даже при кипячении.
Образование в растворе кислот типа Н2[ЭГ6] обуславливает неполноту гидролиза галогенидов ЭГ4. Так, уравнение гидролиза тетрахлорида олова имеет вид:
2 SnCl4 + 4 H2O Ы 2 H2SnCl6 + Sn(OH)4.
Таким образом, гидролизу подвергается лишь одна треть общего количества SnCI4, но гидролиз этой трети идёт до свободного основания, т.е. протекает практически нацело.
Для германия (как и для кремния) характерны германофтористоводородная кислота (Н2GeF6) и её соли. Например, термически устойчивый К2GeF6 (т. пл. 730, т. кип. 835 °С). Германийтетрахлорид не проявляет кислотных свойств в жидком хлористом водороде.
В виде кристаллогидратов были выделены и некоторые свободные комплексные кислоты, например, Н2SnCl6·6H2O (т. пл. 20 °С). Для Sn и Pb известны также производные кислот типа Н4[ЭF8], например, (NH4)4SnF8. Однако было установлено, что в некоторых из этих соединений истинное координационное число центрального атома равно не восьми, а лишь шести. Так, кристаллы кислых солей типа К3НЭF8 слагаются из ионов К+, ЭF62- и HF2-. Вместе с тем и для германия и для олова были получены кристаллические производные состава 4 ХеF6·ЭF4, для которых вероятна структура (ХеF5)4ЭF8.
Соли кислородных кислот для четырёхвалентных Ge, Sn и Pb малохарактерны. Получены, в частности, сульфаты Э(SO4)2 и ацетаты Э(CH3COO)4. Все они легко гидролизуются.
Cульфат четырёхвалентного олова Sn(SO4)2 образуется при взаимодействии Sn с горячей крепкой Н2SO4. Из раствора он выделяется в виде бесцветных игл состава Sn(SO4)2·2H2O. Константа диссоциации по схеме
Sn(SO4)2Ы SnSO42+ + SO42-
равна 5·10-3. Жёлтый кристаллический порошок Рb(SO4)2 может быть получен электролизом 80%-ной Н2SO4 со свинцовыми электродами. С сульфатами К, Na и некоторых других металлов он образует жёлтые двойные соли состава М2[Pb(SO4)3]. Водой Рb(SO4)2 полностью гидролизуется с выделением РbO2. Аналогичный гидролиз претерпевает Ge(SO4)2, который может быть получен нагреванием смеси GeCI4 c SO3 в запаянной трубке.
Взаимодействием SnCl4 c N2O5 был получен нитрат четырёхвалентного олова — Sn(NO3)4. Он представляет собой прозрачные кристаллы (т. пл. 91 °С), способные возгоняться в вакууме. Водой Sn(NO3)4 тотчас гидролизуется, в ССl4 растворяется без разложения, а углеводороды окисляет. Также используемая для получения этой соли реакция по схеме
SnCl4 + 4 ClNO3 = 4 Cl2 + Sn(NO3)4
интересна как пример взаимодействия разно поляризованных (отрицательно в SnCl4 и положительно вClNO3) атомов хлора.
Тетраацетат свинца Pb(CH3COO)4 образуется при действии тёплой уксусной кислоты и хлора на сурик по реакции:
Pb3O4 + 8 CH3COOH + Cl2 = PbCl2Ї + 2 Pb(CH3COO)4 + 4 H2O.
При охлаждении раствора Pb(CH3COO)4 кристаллизуется в виде белых игл (т. пл. 175 °С). Подобный же характер имеют кристаллы Ge(CH3COO)4 (т. пл. 156 °С) и Sn(CH3COO)4 (т. пл. 253 °С). Для четырёхвалентного свинца известны соли ряда органических кислот.
Производные четырёхвалентного свинца являются исключительно сильными окислителями (в кислой среде). Так, при кипячении с 30%-ной серной кислотой РbO2 окисляет двухвалентный Мn до марганцовой кислоты, несмотря на то, что последняя сама является очень сильным окислителем. Реакция идёт по уравнению:
5 PbO2 + 2 MnSO4 + 3 H2SO4 = 5 PbSO4 + 2 HMnO4 + 2 H2O.
На окислительных свойствах четырёхвалентного свинца основана работа свинцового аккумулятора.
Свинцовый аккумулятор составляется из решетчатых свинцовых пластин, заполненных пастой из PbO и воды и опущенных в 30%-ную серную кислоту (с плотностью 1,2 г/см3). По реакции
PbO + H2SO4 = PbSO4 + H2O
на поверхности пластин образуется слой труднорастворимого сернокислого свинца. Если теперь через всю систему пропускать постоянный электрический ток в определённом направлении, то у пластин идут следующие реакции (процессы при зарядке):
отрицательный электрод положительный электрод
PbSO4 + 2 e- + 2 H• = Pb + H2SO4 PbSO4 + SO4”- 2 e- = Pb(SO4)2
(Pb•• + 2 e- = Pb) (Pb••- 2 e- = Pb••••)
Pb(SO4)2 + 2 H2O Ы PbO2 + 2 H2SO4
Таким образом, при зарядке аккумулятора отрицательные пластины превращаются в губчатую массу металлического свинца, положительные — в PbO2, а концентрация серной кислоты в растворе повышается.
Если оба электрода не соединены друг с другом проводником, аккумулятор может в заряженном виде сохраняться весьма долго. Напротив, при включении их в цепь через последнюю начинает идти электрический ток в обратном направлении. Возникновение тока обусловлено следующими реакциями у электродов (процессы при разрядке):
отрицательный электрод положительный электрод
Pb + SO4” = PbSO4 + 2 e- PbO2 + 2 H2SO4Ы Pb(SO4)2 + 2 H2O
(Pb = Pb•• + 2 e-) Pb(SO4)2 + 2 e- + 2 H• = PbSO4 + H2SO4
(Pb•••• + 2 e- = Pb••)
Процессы эти обратны имеющим место при зарядке аккумулятора. Получаемый при разрядке свинцового аккумулятора электрический ток имеет напряжение около 2 В. Соединением ряда таких аккумуляторов друг с другом могут быть образованы батареи, достаточно мощные для обеспечения работы электровозов и т. д.
Интересна реакция плюмбодиоксида с хлорноватистой кислотой, протекающая по схеме:
2 PbO2 + 4 HOCl = 2 PbCl2 + 2 H2O + 3 O2.
В щелочной среде окислительные свойства PbO2 проявляются лишь под действием веществ, способных достаточно легко окисляться. Примером может служить реакция по уравнению:
2 Cr(OH)3 + 3 PbO2 + 10 KOH = 3 K2PbO2 + 2 K2CrO4 + 8 H2O.
В противоположность галогенидам ЭГ4 галогенпроизводные двухвалентных Sn и Pb имеют отчётливо выраженный характер солей. Все они хорошо кристаллизуются, плавятся лишь при сравнительно высоких температурах и подвергаются в растворе значительно меньшему гидролизу, чем соответствующие галогениды ЭГ4. Несколько ближе к последним по свойствам малоустойчивые галогениды двухвалентного германия.
В парах SnF2, помимо мономеров, обнаружено наличие димеров и тримеров, а плюмбофторид имеет в парах тенденцию к дисмутации по схеме:
2 PbF2 = PbF4 + Pb.
Галогениды олова хорошо растворимы в воде (кроме SnI2), галогениды свинца — плохо. По ряду Сl-Br-I растворимость и тех и других уменьшается.
Расплавом безводного SnCl2 пользуются иногда для освобождения чернового олова от свинца (по реакции: SnCl2 + Pb = PbCl2 + Sn). Оно растворимо в ацетоне (приблизительно 1:2 по массе) и некоторых других органических растворителях (спирт, эфир), а из водных растворов выделяется в виде бесцветного, плавящегося при 40 °С кристаллогидрата SnCl2·2H2O (“оловянная соль”). Фтористое олово является одной из наиболее эффективных фторирующих добавок в зубные пасты.
Общим способом образования галогенидов двухвалентного германия является реакция по схеме:
GeГ4 + Ge = 2 GeГ2.
Галогениды германия представляют собой бесцветные (кроме жёлтого GеI2) твёрдые вещества, весьма склонные к дисмутации на GeГ4 и Ge. По ряду F-Cl-Br-I устойчивость возрастает. Водой они очень сильно гидролизуются.
Термическим разложением GeCI4 около 1000 °С был получен коричневый (после очистки — жёлтый) субхлорид германия состава GeCl (точнее, GeCl0,9). Это микрокристаллическое вещество устойчиво в вакууме до 360 °С, а при дальнейшем нагревании подвергается дисмутации на Ge и GeCl4.
Частичное образование аналогичных субгалогенидов Sn и Pb является вероятной причиной растворимости этих металлов в их расплавленных галогенидах ЭГ2. Такая растворимость возрастает по ряду галогенидов Сl-Br-I и при повышении температуры.
Подобно ЭГ4, двухвалентные галогениды Ge, Sn и Pb способны образовывать комплексные соединения, которые, однако, значительно менее устойчивы. Характерны для них комплексы типов M[ЭГ3] и M2[ЭГ4]. В разбавленных растворах все они почти нацело разложены на соответствующие простые ионы. Напротив, в более крепких растворах (или при избытке иона Г-) образуется заметное количество комплексных ионов. Этим обусловлена лучшая растворимость галогенидов свинца в крепких растворах галогеноводородных кислот или их солей по сравнению с чистой водой. По структуре интересна двойная соль состава 2SnF2·NaF. Её кристаллы содержат анионы [F(SnF2)2]-с фторидными мостиками [d(FSn) = 222 пм] между двумя ионами SnF2 [d(SnF) = 207 пм].
Почти бесцветный в безводном состоянии КPbI3 (т. пл. 349 °С), или раствор его в ацетоне, является чувствительным реактивом на влагу, так как под действием воды он тотчас желтеет вследствие разложения с выделением PbI2. Константа нестойкости [PbI3]” равна 2·10-6.
В связи с ослаблением основных свойств по ряду гидрокcидов Pb(OH)2-Sn(OH)2-Ge(OH)2 гидролиз производящихся от них солей по этому ряду увеличивается: в то время как соли двухвалентного Pb гидролизованы незначительно, производные двухвалентного Ge в разбавленных растворах разлагаются водой почти нацело. Соли Sn2+ обладают промежуточными свойствами.
Большинство солей Sn2+ бесцветно и хорошо растворимо в воде. Производные двухвалентного олова (в ещё большей степени — германия) являются сильными восстановителями. Растворы их постепенно окисляются уже кислородом воздуха.
Наибольшее практическое значение из солей Sn2+ имеетхлористое олово (SnCI2). Применяется оно главным образом как восстановитель. Например, соли ртути восстанавливаются им до металла:
HgCl2 + SnCl2 = SnCl4 + Hg.
Cоли кислородных кислот для двухвалентного олова (и германия) малохарактерны. Из них SnSO4 используется при электролитическом лужении (т. е. покрытии других металлов оловом).
Соли двухвалентного свинца восстановителями не являются. Большинство их бесцветно и малорастворимо в воде. Из часто встречающихся в практике хорошо растворяются только азотнокислая Pb(NO3)2 и уксуснокислая Pb(CH3COO)2 соли.
Белые игольчатые кристаллы SnSO4 хорошо растворимы в воде (около 1:2 по массе). Их термическое разложение по схеме SnSO4 = SnO2 + SO2 идёт (в атмосфере азота) выше 360 °С. Термическое разложение оксалата олова по схеме:
SnC2O4 = CO2 + CO + SnO
может служить методом получения его оксида.
Нитрат и ацетат свинца (свинцовый сахар — Pb(CH3COO)2·3H2O, т. пл. 58 °С) получают обычно растворением свинца в соответствующих кислотах. Первая из этих солей применяется главным образом как исходный материал для получения других соединений Pb, вторая — в красильном деле и медицине (“свинцовая примочка” и др.). Нитрат свинца в растворе довольно сильно диссоциирован (константа диссоциации иона PbNO3• равна 0,7), а молекула Pb(CH3COO)2 малодиссоциирована (К1 = 3·10-2, К2 = 4·10-3). Пропитанная раствором ацетата свинца и затем высушенная бумага при поджигании не горит, а тлеет, как трут. Расплавленный PbCl2 обладает значительной электропроводностью, а при застывании образует роговидную массу (“роговой свинец”).
На галогениды свинца похожи по свойствам бесцветные Pb(CN)2 и Pb(NCS)2. Очень малая растворимость в воде PbI2, PbSO4 и PbCrO4 используется при химических анализах. Хромовокислый свинец применяется также в качестве жёлтой минеральной краски (“хромовая жёлтая”). Цианамид свинца PbNCN находит использование в составах для антикоррозионных покрытий. При нагревании выше 250 °С (в отсутствие воздуха) соль эта разлагается по схеме:
2 PbNCN = 2 Pb + (CN)2 + N2.
При медленном охлаждении горячего насыщенного (лучше слегка подкисленного) раствора PbI2 соль эта выделяется в виде очень красивых золотистых листочков. Йодистый свинец светочувствителен: во влажном воздухе он постепенно разлагается на свету с образованием PbO и I2.
Практически важной основной солью двухвалентного свинца долгое время был карбонат приблизительного состава 2PbCO3·Pb(OH)2, служащий для изготовления белой масляной краски — свинцовых белил. Последние применялись как самостоятельно, так и в смеси с другими красками. Процесс получения основного карбоната свинца детально описан в “Трактате о камнях” Теофраста (315 г. н. э.). Имеется указание на возможность использования этого вещества как исходного сырья для производства искусственного перламутра.
Достоинством свинцовых белил является их большая кроющая способность, серьёзным недостатком — постепенное потемнение окрашенных предметов на содержащем следы Н2S воздухе (каков, в частности, воздух городов) вследствие перехода белого основного карбоната в чёрный PbS. Из-за ядовитости свинцовых белил применение их в настоящее время запрещено.
Как свинцовые белила, так и другие масляные краски приготовляются путём растирания тех или иных окрашенных твёрдых веществ с высыхающими на воздухе растительными маслами (обычно — льняным или конопляным). Высыхание этих масел обусловлено их окислением кислородом воздуха. Оно значительно ускоряется, если в масле присутствуют небольшие количества некоторых оксидов (PbO, MnO2 и др.), служащих катализаторами. Содержащее такие оксиды (“сиккативы”) высыхающее растительное масло называется олифой.
Приготовленная на олифе цветная масляная краска, кроме придающих ей ту или иную окраску веществ (“пигментов”), всегда содержит какой-либо тонкий белый порошок, сообщающий краске непрозрачность и не допускающий образования пор при высыхании масла. Такой “основой” может служить основной карбонат свинца. Он придаёт краске большую кроющую способность, что позволяет довольствоваться нанесением на предмет очень тонкого её слоя.
Почти все картины старых мастеров писаны красками, приготовленными на основе свинцовых белил. Вследствие потемнения с течением времени многие из этих картин уже утратили первоначальные оттенки. Последние часто могут быть восстановлены путём осторожной обработки картин разбавленным раствором перекиси водорода, так как под её действием чёрный PbS переходит в белый PbSO4, почти не отличающийся по цвету от основного карбоната свинца.
Отвечающие типам ЭS и ЭS2 сульфиды могут быть получены (кроме PbS2) как сухим путём (из элементов), так и действием сероводорода на содержащие ионы Э••или Э••••растворы соответствующих солей. В последнем случае образуются осадки следующих цветов:
GeS2 SnS2 GeS SnS PbS
белый жёлтый буро-красный бурый чёрный
В воде и разбавленных кислотах эти сульфиды практически нерастворимы. Исключение представляет GeS2, слегка растворимый в воде и гидролитически разлагающийся ею.
Сульфиды типов ЭS и ЭS2 существенно отличаются друг от друга по своему отношению к сернистому аммонию. В то время как на первые он не действует, вторые переводятся им в раствор с образованием аммонийных солей тиогерманиевой (H2GeS3) и тиооловянной (H2SnS3) кислот по схеме:
(NH4)2S + ЭS2 = (NH4)2ЭS3
Ввиду неустойчивости этих кислот в свободном состоянии при подкислении растворов их солей происходит отщепление Н2S и осаждение сульфида ЭS2.
Кристаллы PbS (т. пл. 1114 °С) имеют решётку типа NaCl. Подобно металлическому германию, вещество это интенсивно поглощает энергию в световом и близких к нему диапазонах, но практически прозрачно для теплового излучения. Аналоги сернистого свинца — PbSe (т. пл. 1065 °С) и PbTe (т. пл. 924 °С) — обладают полупроводниковыми свойствами, причём селенид свинца очень чувствителен к инфракрасным лучам.
Непосредственное применение из рассмотренных сульфидов находит главным образом кристаллическое SnS2, порошок которого под названием “муссивного золота” входит в состав красок для золочения. Выработку его ведут обычно путём постепенного нагревания до 300 °С смеси амальгамы олова с серным цветом и NH4Cl, причём SnS2 получается в виде золотисто-жёлтых пластинок. Наиболее древнее дошедшее до нас описание муссивного золота содержится в сочинениях китайского химика Ко Хуна.
Соединения с азотом из всех элементов рассматриваемой подгруппы наиболее характерны для германия. Его серый нитрид (Ge3N4) может быть получен действием NH3 на металлический германий (или GeO2) при 700 °С. Вода, щёлочи и разбавленные кислоты на нитрид германия не действуют, а распад его на элементы идёт лишь около 800 °С. Аналогичный по составу коричневый нитрид олова (Sn3N4) распадаются на элементы уже при 360 °С.
Помимо описанного выше, для Ge (и Sn) известен нитрид состава Ge3N2, являющийся производным двухвалентного германия. Он представляет собой тёмно-коричневый порошок, легко подвергающийся гидролизу. Распад Ge3N2 на элементы начинается около 500 °С.
Нитриды Pb неизвестны. Оранжево-красный имид свинца PbNH может быть получен взаимодействием Pb(NO3)2 и KNH2 в жидком аммиаке. Вещество это крайне неустойчиво и легко взрывается при нагревании или контакте с жидкой водой. Водяным паром оно разлагается на Pb(OH)2 и аммиак.
Несколько особняком в химии Ge, Sn и Pb стоят ихводородные соединения. Для двухвалентных элементов они не характерны, а для четырёхвалентных устойчивость их по ряду Ge-Sn-Pb уменьшается настолько быстро, что существование PbH4 доказано, но свойства его не изучены. Все три гидрида образуются как незначительные примеси к водороду при разложении кислотами сплавов этих элементов с магнием. От водорода они могут быть отделены охлаждением смеси газов жидким воздухом.
Пространственная структура гидридов ЭН4 отвечает тетраэдру с атомом Э в центре. По физическим свойствам GeH4 и SnH4 похожи на аналогичные соединения Si и С. Они также представляют собой бесцветные газы с низкими температурами плавления и кипения, как это видно из приводимого ниже сопоставления:
СН4 | SiH4 | GeH4 | SnH4 | |
| Теплота образования, кДж/моль | +75 | -33 | -92 | -163 |
| d(ЭН), пм | 109 | 148 | 153 | 170 |
Энергии связи Э-Н, кДж/моль | 414 | 318 | 309 | 297 |
Температура плавления, °С | -184 | -185 | -166 | -146 |
Температура кипения, °С | -161 | -112 | -88 | -52 |
При хранении гидриды германия и олова постепенно разлагаются на элементы. Быстро такой распад GeH4 идёт около 350 °С, а SnH4 — уже около 150 °С. Вода, а также разбавленные растворы кислот и щелочей разлагают их сравнительно медленно. Оба гидрида по ядовитости близки к мышьяковистому водороду.
Образование станнометана (SnH4) может производиться в жестяных консервных банках за счёт действия на их полуду органических кислот содержимого. Возможно, что с этим связаны имеющие иногда место случаи тяжёлых отравлений при употреблении в пищу давно изготовленных консервов. Предельно допустимое содержание в них олова составляет 0,02 %.
Из гомологов SnH4 в очень небольших количествах был получен лишь крайне неустойчивый Sn2H6, но свойства его не описаны. Моногерман (GeH4) может быть получен обработкой Мg2Ge раствором бромистого аммония в жидком аммиаке. По отношению к растворам кислот и щелочей он значительнее устойчивее силана. Реакция термического разложения моногермана, как и SnH4, является аутокаталитической. Однако энергия её активации гораздо больше, чем у олова (213 в объёме и 171 кДж/моль на германии). Поэтому с заметной скоростью реакция протекает лишь при повышенных температурах (примерно с 220 °С). Термическим разложением моногермана могут быть получены тонкие плёнки германия на стекле и других изоляторах, что используется при изготовлении высокоомных электрических сопротивлений.
В отличие от СН4 и SiH4 моногерман сравнительно легко образует продукты замещения водорода на металл. Так, действием GeH4 на раствор металлического натрия (или калия) в жидком аммиаке может быть получен натрийгерманил — NaGeH3. Он представляет собой твёрдое вещество, хорошо растворимое в жидком аммиаке с частичной диссоциацией на Na+и GeH3-. При -33 °С натрийгерманил постепенно желтеет, а дальнейшее его нагревание вызывает распад по схеме:
2 NaGeH3 = 2 NaGe + 3 H2.
В форме желтовато-серого аммиаката LiGeH3·2NH3 получено и аналогичное производное лития.
Из других реакций замещения атомов водорода GeH4 на металл интересно взаимодействие его с раствором AgNO3 протекающее по уравнению:
GeH4 + 4 AgNO3 = GeAg4 + 4 HNO3.
Раствор AgNO3 разлагает и SnH4. Разрушение последнего быстро протекает также при соприкосновении его с твёрдыми щелочами и концентрированной Н2SO4.
Будучи по атомной структуре непосредственными аналогами С и Si, элементы подгруппы германия дают в общем соединения тех же типов. Однако свойства этих соединений более или менее закономерно изменяются в связи с изменением химического характера самих элементов.
В частности, по ряду С-Pb уменьшается энергия связей Э-Э: 347 (С-С), 222 (Si-Si), 188(Ge-Ge), 155 кДж/моль(Sn-Sn). С другой стороны, по тому же ряду увеличиваются координационные числа элементов. Например, у фтористых соединений максимальное координационное число углерода составляет четыре (в СF4), кремния и германия — шесть (в солях Н2ЭF6). По отношению к более объёмистым галогенам максимальное координационное число кремния (и углерода) не превышает четырёх, у Ge оно возрастает до шести только для хлора, а у Sn и Pb - даже для иода. Как уменьшение устойчивости связей Э-Э, так и повышение координационного числа по ряду С-Pb обусловлены увеличением в том же ряду размеров соответствующих атомов и ионов.
Одинаковость значений валентности и координационного числа углерода имеет большое значение для химии его соединений, так как ведёт к повышению химической устойчивости многих из них. Последнее стоит в связи с тем обстоятельством, что при химических процессах (особенно между молекулами с малополярными связями) первой стадией часто является присоединение одной из реагирующих частиц к другой и лишь вслед за тем идёт обмен атомами (или ионами) с образованием новых соединений. Очевидно, что в тех случаях, когда координационное число элемента совпадает с его валентностью, внутренняя сфера уже заполнена и присоединение к центральному атому какой-либо посторонней молекулы (или иона) затруднено. Комплексообразователь оказывается экранированным, т.е. как бы “защищённым” окружающими его атомами от внешних воздействий, что и ведёт к медленности протекания всего процесса в целом или даже к практически полному его отсутствию, несмотря на то, что по сути дела он должен был бы иметь место. Именно так следует, по-видимому, понимать многие характерные отличия соединений углерода от аналогичных им производных Si, например большую устойчивость ССl4 по отношению к другим реактивам, легко разлагающим SiCl4. Несомненно, что и химическая инертность насыщенных углеводородов в известной степени обусловлена равенством валентности и координационного числа углерода.
XIV. ЭЛЕМЕНТЫ ТРИАД
Входящие в триады 9 элементов середин больших периодов ранее объединяли под названием VIII группа периодической системы. Это было неудачно. Во-первых, такая VIII группа принципиально отличалась от всех остальных по своей структуре, так как не имела аналогов в малых периодах. Во-вторых, название VIII группы естественно должно принадлежать заканчивающим периоды элементам, т. е. инертным газам.
Исходя из наличия во внешних слоях всех рассматриваемых атомов не более двух электронов, можно ожидать, что тенденция к их дальнейшему присоединению не будет для этих атомов характерна. Элементы триад должны, следовательно, иметь только металлический характер.
Опыт показывает, что по свойствам Fe, Co и Ni довольно близки друг к другу и существенно отличаются от остальных шести элементов. Ввиду эторо представляется рациональным совместное рассмотрение Fe, Co и Ni как членов семейства железа. С другой стороны, сходство многих свойств Ru-Rh-Pd и Os-Ir-Pt позволяет объединить их в семейство платиновых металлов.
1. Семейство железа. Из всех членов
32
Воздух. Кислород.
Воздух.
Сами того не замечая, мы живём на дне огромного воздушного океана. Та смесь газов, которая образует атмосферу, необходима для нас более, чем что-либо другое. Человек может прожить несколько недель без пищи, несколько дней без воды, но не может прожить и нескольких минут без воздуха. В воздухе таятся огромные, пока почти неиспользованные запасы энергии: вследствие неодинакового поглощения солнечных лучей различными участками земной поверхности создаётся неравномерный нагрев воздуха и возникают ветры, за счёт которых могут быть получены многие миллиарды киловатт-часов электроэнергии.
Общая масса атмосферы равна 5,2·1015 т, т.е. составляет менее одной миллионной от массы всего земного шара (6,0·1021 т). Однако на долю каждого человека всё же приходится более 1,5 млн. т воздуха. Около 90% массы атмосферы заключено в слое высотой до 16 км и лишь одна миллионная — выше 100 км.
В древности воздух считался индивидуальным веществом. По учению греческого философа Анаксимена, воздух является началом всего сущего, а позднее он стал рассматриваться в качестве одного из основных элементов природы. То обстоятельство, что воздух имеет массу, было известно уже Аристотелю.
Александрийский учёный Герон (62-150 гг. н.э.) писал о воздухе следующее: “Сосуды, которые кажутся большинству людей пустыми, на самом деле не пусты, а наполнены воздухом… Воздух образован частицами маленькими и лёгкими, в своём большинстве невидимыми… Отсюда должно быть принято, что воздух материален. Приведённый в движение, он становится ветром (так как ветер есть не что иное, как воздух в движении)”.
Первые указания на сложность состава воздуха в сочинениях древних китайских химиков. Из европейцев такое мнение впервые высказал Леонардо да Винчи (конец XV века). Оно было подтверждено опытным путём и стало общепринятым лишь около XVIII века.
Сила ветра измеряется специальными приборами (анемометрами) и обычно оценивается по 12-бальной шкале. Тихий ветер (1) лишь отклоняет дым из трубы, при сильном (6) качаются верхушки деревьев, а ураган (12) причиняет большие разрушения.
Состав воздуха: основные составные части можно подразделить на три группы: постоянные, переменные и случайные. К первым относятся кислород (около 21% по объёму), азот (около 78%) и так называемые инертные газы (около 1%). Содержание этих составных частей практически не зависит от того, в каком месте земного шара взята проба сухого воздуха. Ко второй группе относятся углекислый газ (0,02-0,04%) и водяной пар (до 3%). Содержание случайных составных частей зависит от местных условий: вблизи металлургических заводов к воздуху часто бывают примешаны заметные количества сернистого газа, в местах, где происходит распад органических остатков, — аммиака и т.д. Помимо различных газов, воздух всегда содержит большее или меньшее количество пыли.
Кроме перечисленных газов воздух постоянно содержат следы (т.е. ничтожные количества) озона, водорода, метана, аммиака, оксидов азота и угарного газа. По мере совершенствования методов газового анализа число таких, практически незаметных составных частей воздуха постепенно возрастает.
Атмосферная пыль содержит частицы диаметром от 10-7 до 10-2 см (из которых наиболее мелкие не оседают даже в неподвижном воздухе). Помимо пылинок, возникающих на земной поверхности (частиц почвы, дыма, пыльцы растений и т.д.), некоторое значение имеют пылинки вулканического и даже космического происхождения. Подсчитано, что на Земле ежегодно оседает около 5 млн. т космической пыли. Так как поверхность Земли равна 510 млн. км2, это составляет лишь сотую долю грамма на квадратный метр.
Абсолютная запылённость воздуха может быть в отдельных местах очень различной. Его относительная запылённость быстро уменьшается с высотой.
| Высота, км | 0,1 | 1 | 2 | 3 | 4 | 5 | 6 |
Число пылинок в 1 см3 | 45000 | 6000 | 700 | 200 | 100 | 50 | 20 |
Кубический сантиметр комнатного воздуха обычно содержит миллионы пылинок.
Общая запылённость воздуха, по-видимому, возрастает. Так, было установлено, что за десятилетие с 1957 по 1967 г. помутнение атмосферы над Тихим океаном увеличилось на 30%. Количество пыли, выпадающей в большом городе, огромно. Было подсчитано, что на каждый м2 в Нью-Йорке ежемесячно выпадает до 17 г пыли, а в Токио — даже вдвое больше. Каждый кубический сантиметр воздуха городов содержит несколько тысяч микроорганизмов.
Освобождение от пыли является первой стадией получения кондиционированного воздуха, который, помимо чистоты, характеризуется постоянными температурой и влажностью. Кондиционирование воздуха важно для некоторых отраслей промышленности, а также картинных галерей, музеев и т.д.
Находящийся над Землёй воздух давит на неё с силой более одного килограмма на каждый квадратный сантиметр поверхности. Эту величину легко подсчитать, зная, что нормальное атмосферное давление уравновешивается столбом ртути (плотность 13,6 г/см3) высотой 760 мм. Общее давление атмосферы может быть разложено на давления отдельных составляющих её газов — в этом случае говорят об их парциальных (частичных) давлениях. Например, из общей величины в 760 мм рт. ст. на долю кислорода приходится 760·21/100 = 160 мм рт. ст. Вся жизнь на земной поверхности развивалась в условиях давления атмосферы, поэтому мы не замечаем его, подобно тому как глубоководные рыбы не замечают колоссальных давлений на больших глубинах океана, тогда как на глубине 11 км давление превышает 1000 атм. Среднее атмосферное давление в зависимости от высоты над уровнем моря имеет следующие значения:
| Высота, км | 1 | 2 | 3 | 4 | 5 | 10 | 20 | 50 | 100 | |
| Давление, мм рт. ст. | 760 | 673 | 594 | 524 | 461 | 405 | 210 | 42,0 | 0,76 | 0,0006 |
Соотношение между постоянными составными частями воздуха в нижних слоях атмосферы с высотой почти не меняется.
Налагаемая атмосферным давлением на живые организмы нагрузка гораздо значительнее, чем это представляется с первого взгляда. Общая поверхность человеческого тела составляет в среднем около 20 тыс. см2. Это значит, что человек незаметно для себя испытывает постоянную нагрузку в размере примерно 20 т.
Непосредственно примыкающий к поверхности Земли слой атмосферы характеризуется довольно закономерным изменением температуры — последняя понижается примерно на 6 град с каждым километром высоты. Слой этот — тропосфера — простирается на высоту около 18 км у экватора и 7 км у полюсов. Между ним и Землёй существует известная разность потенциалов, причём тропосфера заряжена положительно, а земная поверхность отрицательно. Основное значение для поддержания разности потенциалов имеет постоянное поступление в атмосферу множества мельчайших капелек морской воды, срываемых с гребней океанских волн и приобретающих при этом значительный положительный заряд.
Более высокие слои атмосферы принято делить на стратосферу (приблизительно до 40 км), мезосферу (40-80 км), термосферу (80-800 км) и экзосферу (выше 800 км). Границы между этими слоями не являются чёткими и несколько изменяются в зависимости от широты местности, времени года и общего состояния атмосферы. Верхняя граница того или иного слоя носит название соответствующей “паузы”. Так, граница между тропосферой и стратосферой называется тропопаузой. На высотах порядка нескольких тысяч километров экзосфера постепенно переходит в межпланетный газ.
Помимо приведённой выше общей классификации атмосферных слоёв, для некоторых из них применяются другие названия. Так, слой высотой 30-80 км, в котором преимущественно протекают химические реакции под действием солнечных лучей, иногда называют хемосферой, слой выше 80 км, характеризующийся большим относительным содержанием заряженных частиц — ионосферой. Под “верхней атмосферой” в различных случаях понимают разные слои атмосферы.
Основной химический состав атмосферы примерно до 1000 км остаётся азотно-кислородным. В противоположность монотонно уменьшающемуся давлению, температурная кривая имеет минимум на высоте около 20 км, максимум около 50 км и новый минимум в мезопаузе. После этого температура начинает расти, достигая примерно 900 °С уже на высоте 200 км.
Общий характер высотного изменения температуры воздуха был предугадан Аристотелем. Он делил атмосферу на три слоя, из которых прилегающий к Земле пригоден для жизни, следующий сильно охлаждён, а самый верхний, наоборот, сильно нагрет. Средний молекулярный вес воздуха 29.
При достаточном охлаждении воздух переходит в жидкое состояние. Жидкий воздух можно довольно долго сохранять в сосудах с двойными стенками, из пространства между которыми для уменьшения теплопередачи выкачан воздух. Подобные сосуды используются, например, в термосах.
Свободно испаряющийся при обычных условиях жидкий воздух имеет температуру около –190 °С. Состав его непостоянен, так как азот улетучивается быстрее кислорода. По мере удаления азота цвет жидкого воздуха изменяется от голубоватого до бледно-синего (цвет жидкого кислорода).
До ХIX века считали, что газы являются таковыми по самой своей природе, и вопрос о их сжижении даже не возникал. Лишь в 20-х годах ХIХ века, применяя значительные давления, удалось получить в жидком состоянии хлор, аммиак, диоксид углерода и ряд других веществ “газообразной природы”. Однако оставались ещё многие, в частности основные газы воздуха — кислород и азот, которые, несмотря на все усилия, не сжимались. На них перенесли то представление, которое раньше было общим, и стали считать их “постоянными” газами. Только в 1877 г. впервые удалось получить в жидком состоянии одни из этих “постоянных” газов — кислород. Вслед за тем были сжиженны и все другие.
Причина неудач ранних попыток сжижения газов лежала в том, что ещё неясна была сущность различия между газообразным и жидким состоянием вещества. Мы знаем теперь, что в обоих случаях имеет место и взаимное притяжение молекул, и их взаимное расталкивание. Жидкое состояние вещества характеризуется преобладанием первого, газообразное — второго. Взаимное притяжение молекул практически не зависит от температуры. Напротив, обусловленное их ударами друг о друга взаимное расталкивание весьма сильно зависит от температуры, так как её величина определяет скорость движения молекул и их кинетическую энергию. Газ может быть переведён в жидкое состояние лишь тогда, когда стяжение получает преобладание над расталкиванием или, по крайней мере, становится равным ему. Та температура, при которой расталкивание уравновешивается притяжением, характеризуется отсутствием различия между жидкостью и её паром и называется критической. Существование такой температуры было впервые установлено Д. И. Менделеевым (1961 г.).
Критическая температура различна для различных веществ и, например, для хлора равна +144 °С. Поэтому, применив достаточное давление, хлор можно перевести в жидкое состояние и без его охлаждения. Критические температуры основных газов воздуха лежат, наоборот, очень низко: кислорода при –118 °С и азота при –147 °С. Поэтому воздух можно перевести в жидкое состояние, лишь охладив его предварительно ниже указанных температур. Между тем исследователи раннего периода пытались получить жидкий воздух, применяя высокие давления, но не заботясь о достаточном охлаждении.
Наиболее простое экспериментальное определение критической температуры жидкостей производят следующим образом. В толстостенной стеклянной трубке запаивают небольшое количество исследуемого вещества. На границе раздела жидкости и её пара образуется мениск. При постепенном нагревании трубки в ней всё время увеличивается давление, поэтому жидкость целиком не испаряется и мениск отчётливо виден. Вблизи критической температуры он становится всё более плоским и, наконец, исчезает. Та температура, при которой происходит исчезновение мениска (т.е. поверхности раздела двух фаз), и является критической температурой исследуемого вещества.
Схема получения жидкого воздуха заключается в том, что предварительно освобождённый от пыли, влаги и углекислого газа воздух сжимается компрессором до 200-250 атм (при одновременном охлаждении водой), проходит первый теплообменник и затем разделяется на два потока. Большая часть направляется в детандер — поршневую машину, работающую за счёт расширения воздуха. Последний, значительно охладившись в детандере, омывает оба теплообменника и, охладив текущий навстречу сжатый воздух, покидает установку. Другой поток сжатого воздуха, охлаждённый ещё более во втором теплообменнике направляется в расширительную камеру, после чего покидает установку вместе с воздухом из детандера. Вскоре наступает момент, когда в расширительной камере достигается температура сжижения, и затем он уже непрерывно получается в жидком состоянии.
В 1938 г. П.Л. Капицей был разработан метод получения жидкого воздуха при низком давлении — всего 5-6 атм.
По мере испарения азота жидкий воздух обогащается кислородом, причём температура его кипения постепенно повышается. Одновременно возрастает и плотность жидкого воздуха (приблизительно 0,94 г/см3 для нормального состава). Температура его затвердевания также зависит от состава, причём наинизшая она (– 223 °С) при содержании 78% кислорода.
При температурах жидкого воздуха свойства многих веществ резко изменяются. Например, жёлтая при обычных условиях сера становится белой, Такие жидкости и газы, как спирт, диоксид углерода и т.п., при соприкосновении с жидким воздухом затвердевают. Свинцовая пластинка после погружения в жидкий воздух издаёт при ударе ясный металлический звон, резина становится настолько хрупкой, что при ударе разбивается на куски, и т.д.
Химические реакции при температуре жидкого воздуха вообще очень сильно замедляются. Однако благодаря большой концентрации в нём кислорода (концентрацией называется количество вещества в единице объёма или массы), смешанные с жидким воздухом горючие вещества горят гораздо энергичнее, чем в обычных условиях. Например, смоченная жидким воздухом вата сгорает со вспышкой подобно бездымному пороху.
На этом основано применение жидкого воздуха для взрывных работ в горном деле, где используются патроны с пропитанными им горючими материалами. Подобное взрывчатое вещество (оксиликвит) посиле взрыва лишь немногим уступает динамиту, при этом оно дешевле и безопасней в обращении. Ещё эффективнее оксиликвиты на основе жидкого кислорода.
Инертные газы.
В 1893 г. было обращено внимание на несовпадение плотностей азота из воздуха и азота, получаемого при разложении азотных соединений: литр азота из воздуха имел массу 1,257 г, а полученный химическим путём — 1,251 г. Произведённое для выяснения этого загадочного обстоятельства очень точное изучение состава воздуха показало, что после удаления всего кислорода и азота получался небольшой остаток (около 1%), который ни с чем химически не реагировал. Открытие нового элемента, называемого аргоном (по-гречески недеятельный), представило, таким образом, “торжество третьего знака”. Молекулярный вес аргона оказался равным 39,9. Так как молекула его одноатомна, атомный вес аргона равен молекулярному.
Вопрос об атомности молекулы аргона был разрешён при помощи кинетической теории. Согласно последней, количество тепла, которое нужно затратить для нагревания моля газа на один градус, зависит от числа атомов в его молекуле. При постоянном объёме моль одноатомного газа требует 13 Дж газа, двухатомного — 21 Дж. Для аргона опыт давал 13,5 Дж, что и указывало на одноатомность его молекулы. То же относится и к другим инертным газам.
Следующий по времени открытия инертный газ — гелий (“солнечный”) — был обнаружен на Солнце раньше, чем на Земле. Это оказалось возможным благодаря разработанному в пятидесятых годах прошлого века методу спектрального анализа.
Если тонкий пучок “белого” солнечного света направить на стеклянную призму, он разлагается на лучи различных цветов радуги. Каждый луч может быть охарактеризован определённой длиной волны (l) или частотой колебаний (n), т.е. числом волн, сменяющихся за одну секунду.
Греческие буквы l и n читаются соответственно “лямбда” и “ню”. Выражаемые ими величины легко могут быть переведены друг в друга, так как они связаны соотношением: ln = с, где с — скорость света (3·1010 см/с). Отсюда следует, что чем меньше l, тем больше n, и обратно.
Для измерения длин световых волн (и других очень малых длин) обычно применяются следующие единицы: микрон (мк, m) = 0,001 мм = 10-4 см; миллимикрон (ммк, mm) = 0,001 мк = 10-7 см; ангстрем (Г) = 0,1 ммк = 10-8 см.
По международному соглашению (1960 г.) при образовании кратных и дольных единиц рекомендуется использовать определённые приставки к основным единицам (м, с и др.). Ниже приводятся соответствующие множители, названия отвечающих им приставок, их русские и латинские обозначения:
1012 109 106 103 102 101 10-1 10-2 10-3 10-6 10-9 10-12
тера гига мега кило гекто дека деци санти милли микро нано пико
Т Г М к г да д с м мк н п
T G M k h da d c m m n p
В этой системе обозначений 1 микрон = 1 мкм, 1 миллимикрон = 1 нм и 1 ангстрем = 0,1 нм = 100 пм.
По обе стороны от видимого спектра располагаются невидимые лучи: инфракрасные и ультрафиолетовые, которые могут быть обнаружены и изучены при помощи различных физических методов.
Лежащие за пределами видимого спектра лучи обладают рядом интересных особенностей. Ультрафиолетовые лучи при определённых длинах волн обладают сильным бактерицидным (убивающим бактерии), а при несколько больших — эритемным (вызывающим загар кожи) действием. Облучение ими в умеренных дозах благотворно влияет на организм человека. Установлено, что насекомые весьма чувствительны к ультрафиолетовым лучам, которые привлекают их даже сильнее, чем обычный видимый свет.
На долю инфракрасных лучей приходится около 50% всей доходящей до Земли солнечной энергии, и они имеют основное значение для жизни растений. Лучи эти почти не задерживаются туманом, что позволяет, в частности, фотографировать земную поверхность сквозь облачный покров. Инфракрасные лучи испускаются всяким нагретым предметом, в том числе каждым теплокровным животным (характерные длины волн порядка 0,01 мм). Исследованием, проведённым на гремучих змеях, было выяснено, что они имеют в передней части головы специальные теплочувствительные органы и при охоте руководствуются главным образом тепловым излучением своих жертв. Высокочувствительные приёмники в инфракрасном диапазоне улавливают разности температур до тысячных долей градуса. Такое “тепловидение” позволяет решать ряд важных задач — от медицинской диагностики некоторых заболеваний до точного определения местонахождения самолётов в полной темноте.
Если внести в пламя горелки какую-нибудь летучую при нагревании соль натрия, оно окрасится в жёлтый цвет, при внесении летучих соединений меди — в сине-зелёный цвет и т. д. Каждый химический элемент при достаточном нагревании испускает лучи определённых, характерных для него длин волн.
Определение длин световых волн осуществляется с помощью спектроскопа. Прибор этот и дал возможность по спектру Солнца установить его химический состав. Ещё в 1868 г. были таким путём обнаружены линии, не отвечающие ни одному из известных веществ. Эти линии приписали новому элементу — гелию. На Земле он был впервые (1895 г.) найден в газах, выделяющихся при нагревании минерала клевеита.
Через несколько лет после открытия аргона и гелия (в 1898 г.) были выделены из воздуха ещё три инертных газа: неон (“новый”), криптон (“скрытый”) и ксенон (“чуждый”). Насколько трудно было их обнаружить, видно из того, что 1 м3 воздуха, наряду с 9,3 л аргона, содержит лишь 18 мл неона, 5 мл гелия, 1 мл ксенона и 0,09 мл ксенона.
Последний инертный газ — радон — был открыт в 1900 г. при изучении некоторых минералов. Содержание его в атмосфере составляет лишь 6•10-18 % по объёму (что соответствует 1-2 атомам в кубическом сантиметре). Было подсчитано, что вся земная атмосфера содержит лишь 374 л радона.
Для инертных газов характерно полное (He, Ne, Ar) или почти полное (Kr, Xe, Rn) отсутствие химической активности. В периодической системе они образуют особую группу (VIII). Разделение инертных газов основано на различии их химических свойств.
Вскоре после открытия инертных газов образованная ими в периодической системе группа была названа нулевой, чтобы подчеркнуть этим нулевую валентность данных элементов, т. е. отсутствие у них химической активности. Такое название часто применяется и в настоящее время, однако по существу периодического закона правильнее считать группу инертных газов восьмой, так как этими элементами соответствующие периоды не начинаются, а заканчиваются.
Для физической характеристики того или иного вещества наибольшее значение обычно имеет выяснение тех условий, при которых происходит изменение его агрегатного состояния (газообразного, жидкого или твёрдого). В твёрдом виде каждое вещество характеризуется некоторым строго закономерным расположением составляющих его частиц, в газообразном и жидком — более или менее беспорядочным. При последовательном нагревании твёрдого вещества энергия колебательного движения его частиц всё время увеличивается, в результате чего усиливается и их взаимное расталкивание. Рано или поздно достигается такая температура (температура плавления), при которой притяжение частиц друг к другу уже не может обеспечить сохранение строгого порядка в их расположении: вещество плавится.
Однако в жидкости взаимное притяжение молекул ещё достаточно, чтобы удержать их вместе, и лишь отдельным, наиболее быстро в данный момент движущимся молекулам удаётся оторваться от поверхности. При дальнейшем нагревании число таких молекул всё возрастает, т. е. увеличивается давление пара данного вещества. Наконец, достигается для каждого вещества температура (температура кипения), при которой давление его пара становится равным внешнему давлению, парообразование начинает идти не только с поверхности, но и в массе жидкости — последняя “закипает”.
Очевидно, что температура кипения должна сильно зависеть от внешнего давления. Напротив, температура плавления при небольших его количествах заметно не изменяется.
Наиболее практически важно знание тех температурных условий, которые отвечают изменениям агрегатного состояния при нормальном атмосферном давлении (760 мм рт. ст.). Они обычно и указываются как температуры или “точки” плавления (т. пл.) и кипения (т. кип.) рассматриваемого вещества. Значения их для инертных газов видны из приводимого ниже сопоставления:
| Не | Ne | Ar | Kr | Xe | Rn | ||
| Атомный номер | 2 | 10 | 18 | 36 | 54 | 86 | |
| Атомная масса | 4,00260 | 20,179 | 39,948 | 83,80 | 131,3 | 222 | |
Температура плавления, °С | -271 | -249 | -189 | -157 | -112 | -71 | |
Температура кипения, °С | -269 | -246 | -186 | -153 | -108 | -62 |
Твёрдое состояние гелия устойчиво под давлением не ниже 25 атм.
Количество тепла, необходимое для перевода вещества из твёрдого состояния в жидкое, носит название теплоты плавления, а для перевода из жидкого состояния в парообразное — теплоты испарения рассматриваемого вещества. Обе величины относят к переходам, происходящим под нормальным давлением. Для инертных газов они имеют следующие значения (кДж/моль):
| He | Ne | Ar | Kr | Xe | Rn | |
| Теплота плавления | 0,008 | 0,33 | 1,2 | 1,6 | 2,3 | 2,9 |
| Теплота испарения | 0,08 | 1,8 | 6,5 | 9,0 | 12,6 | 16,8 |
Как видно из приведённых данных, теплоты испарения во всех случаях гораздо больше теплот плавления. И те, и другие величины возрастают вместе с повышением температур плавления и кипения инертных газов.
Значения плотности инертных газов в жидком состоянии (при температуре кипения) и их относительные теплопроводности (при 0 °С) равны:
| He | Ne | Ar | Kr | Xe | |
Плотность, г/см3 | 0,13 | 1,2 | 1,4 | 2,6 | 3,1 |
| Относительная теплопроводность (воздух = 1) | 6,0 | 1,96 | 0,73 | 0,38 | 0,22 |
Ниже сопоставлены критические температуры инертных газов и те давления, которые необходимы и достаточны для их перевода при этих температурах из газообразного состояния в жидкое, — критические давления:
| He | Ne | Ar | Kr | Xe | Rn | |
Критическая температура, °С | -268 | -229 | -122 | +64 | -16,6 | +104 |
| Критическое давление, атм | 2,3 | 27 | 48 | 54 | 58 | 62 |
Гелий был последним из газов переведён в жидкое и твёрдое состояние. По отношению к нему имели место особые трудности, обусловленные тем, что в результате расширения при обычных температурах гелий не охлаждается, а нагревается. Лишь ниже 250 °С он начинает вести себя “нормально”. Отсюда следует, что обычный процесс ожижения мог быть применён к гелию лишь после его предварительного очень сильного охлаждения. С другой стороны, и критическая температура гелия лежит крайне низко. В силу этих обстоятельств благоприятные результаты при работе с гелием были получены лишь после овладения методикой оперирования с жидким водородом, пользуясь испарением которого только и можно было охладить гелий до нужных температур. Получить жидкий гелий удалось впервые в 1908 г., твёрдый гелий — в 1926 г. Интересно, что жидкий гелий практически не растворяет никакие другие вещества.
Точки кипения и плавления гелия находятся в непосредственной близости к наинизшему возможному пределу охлаждения вещества — температуре абсолютного, который лежит при -273,15 °С (точно). Хотя абсолютный нуль недостижим, в лабораторных условиях уже были получены температуры, отличающиеся от него лишь на миллионные доли градуса.
От абсолютного нуля начинается отсчёт по шкале абсолютных температур, часто применяемой при научных и технических исследованиях. Абсолютная шкала очень удобна, так как не содержит отрицательных температур. Градус её (К) имеет такую же величину, как и градус обычной шкалы Цельсия (°С). Поэтому соотношение между отсчётами по шкалам абсолютной (Т) и Цельсия (t) даётся простыми выражениями T = t + 273,15 и t = Т - 273,15.
Согласно классической кинетической теории, температура абсолютного нуля характеризуется тем, что при ней прекращается всякое движение частиц, т. е. наступает полный покой. В настоящее время установлено, что частицы вещества сохраняют некоторую колебательную энергию даже при абсолютном нуле. Эта “нулевая энергия” тем больше, чем меньше массы частиц и чем сильнее они взаимодействуют друг с другом. Общая нулевая энергия многоатомных молекул может достигать значительных величин.
Неустойчивость твёрдого состояния гелия под обычным давлением обусловлена крайне малыми силами стяжения между его атомами. Из-за этого уже небольшая сама по себе нулевая энергия гелия (около 210 Дж/моль) оказывается достаточной для нарушения того строгого порядка расположения частиц, который обязателен для твёрдого тела. Повышение давления, искусственно сближая частицы, компенсирует тем самым недостаточность их собственных сил стяжения и поэтому повышает устойчивость твёрдого состояния.
Если точка абсолютного нуля принципиально ограничивает возможности получения низких температур, то для высоких температур подобного принципиального ограничения нет. Чем выше температура, тем больше возможностей для взаимодействия веществ друг с другом и тем быстрее эти взаимодействия протекают. Однако по мере повышения рабочих температур быстро возрастают трудности технического оформления и эксплуатации соответствующих установок. Поэтому практически используемые для проведения химических процессов температуры обычно не превышают 2000 °С.
Для приближённой характеристики высоких температур иногда пользуются указанием на тип свечения нагреваемого вещества (твёрдого или жидкого). Обычно различают области различных яркостей красного (600-1000 °С), жёлтого (1000-1300 °С) или белого (1300-1500 °С) каления.
Очень высокие температуры могут быть получены различными путями. Например, электрическая дуга с водяным охлаждением при диаметре токопроводящего канала 2,4 мм и силе тока 1450 А даёт на оси канала температуру 55000 °С (что примерно в 2,5 раза выше температуры канала молнии). Для измерения столь высоких температур используются методы астрофизики.
Все инертные газы бесцветны и состоят из одноатомных молекул. Растворимость их при переходе от гелия к радону быстро повышается. Так, в 100 объёмах воды растворяется при 0 °С следующее число объёмов инертного газа:
| He | Ne | Ar | Kr | Xe | Rn |
| 1,0 | 2,2 | 5,7 | 11,1 | 24,2 | 41,5 |
Органические растворители (спирт, бензол и др.) дают подобный же ход изменения растворимости, но растворяют инертные газы значительно лучше воды.
Гелий (обычно с добавкой 15% водорода) может быть использован, в частности, для наполнения дирижаблей. Подъёмная сила последних определяется разностью масс воздуха и заполняющего газа, в объёме дирижабля.
Получение гелия в больших количествах стало возможным лишь после открытия источников газов, содержащих гелий. В настоящее время газ этот стал доступен для многих отраслей техники. Весьма перспективна, например, электросварка металлов в атмосфере гелия. Следует отметить, что он способен более или менее быстро проникать сквозь перегородки из стекла, пластмасс и некоторых металлов (но не железа). Хранят его в коричневых баллонах с белой надписью “Гелий”.
Отсутствие у тяжёлых инертных газов полной химической инертности было обнаружено лишь в 1962 г.: оказалось, что они способны соединяться с наиболее активными неметаллом — фтором (и только с ним). Ксенон (и радон) реагируют довольно легко, криптон — гораздо труднее. Получены ХеF2, XeF4, XeF6 и малоустойчивый КrF2. Все они представляют собой бесцветные летучие кристаллические вещества. Лёгкие инертные газы так и останутся полностью инертными.
Инертные газы находят довольно разнообразное практическое применение. В частности, исключительно важна роль гелия при получении низких температур, так как жидкий гелий является самой холодной из всех жидкостей.
Искусственный воздух, в составе которого азот заменён гелием, был впервые применён для обеспечения дыхания водолазов. Растворимость газов с возрастанием давления сильно увеличивается, поэтому у опускающегося в воду и снабжённого обычным воздухом водолаза кровь растворяет азота больше, чем в нормальных условиях. При подъёме, когда давление падает, растворённый азот начинает выделяться и его пузырьки частично закупоривают мелкие кровеносные сосуды, нарушая тем самым нормальное кровообращение и вызывая приступы “кессонной болезни”. Благодаря замене азота гелием болезненные явления резко ослабляются вследствие гораздо меньшей растворимости гелия в крови, что особенно сказывается именно при повышении давлениях. Работа в атмосфере “гелийного” воздуха позволяет водолазам опускаться на большие глубины (свыше 100 м) и значительно удлинять сроки пребывания под водой.
Так как плотность такого воздуха примерно в три раза меньше плотности обыкновенного, дышать им гораздо легче. Этим обусловлено большое медицинское значение гелийного воздуха при лечении астмы, удуший и т.п., когда даже кратковременное облегчение дыхания больного может спасти ему жизнь. Подобный гелийному, “ксеноновый” воздух (80% ксенона, 20% кислорода) оказывает при вдыхании сильное наркотическое действие, что может найти медицинское использование.
Неон и аргон широко используются электротехнической промышленностью. При прохождении электрического тока сквозь заполненные этими газами стеклянные трубки газ начинает светиться, что применяется при оформлении световых надписей и т.п. Расход электроэнергии в таких газосветных трубках очень мал.
Мощные неоновые трубки особенно пригодны для маяков и других сигнальных устройств, так как красный свет мало задерживается туманом. Цвет свечения гелия по мере уменьшения его давления в трубке меняется от розового через жёлтый к зелёному. Для Аr, Kr и Xe характерны различные оттенки голубого цвета.
Аргон (обычно в смеси с 14% азота) служит также для заполнения электроламп. Вследствие значительно меньшей электропроводности ещё лучше подходит для этой цели криптон и ксенон: заполненные ими электролампы дают больше света при том же расходе энергии, лучше выдерживают перегрузку и долговечнее обычных. Атмосферой аргона широко пользуются как защитной при различных химических работах и производственных процессах, когда нужно изолировать реагирующие вещества от окружающего пространства. Хранят аргон в чёрных баллонах с синей надписью “Аргон” и белой полосой над ней.
Кислород.
Кислород является самым распространённым элементом земной коры. В атмосфере его находится около 23 вес.%, в составе воды — около 89 %, в человеческом организме — около 65 %, в песке содержится 53 % кислорода, в глине — 56 % и т.д. Если подсчитать его количество в воздухе (атмосфере), воде (гидросфере) и доступной непосредственному химическому исследованию части твёрдой земной коры (литосфере), то окажется, что на долю кислорода приходится примерно 50 % их общей массы. Свободный кислород содержится почти исключительно в атмосфере, причём количество его оценивается в 1,2•1015 т. При всей громадности этой величины она не превышает 0,0001 общего содержания кислорода в земной коре.
Изучение химических превращений земной коры составляет предмет геохимии. С позиций этой науки значение того или иного элемента для протекающих в земной коре химических взаимодействий определяется его относительным числом атомов. Поэтому более правильным является сопоставление распространённости отдельных элементов не в весовых, а в атомных процентах. Последние находят, деля весовые проценты на соответствующие атомные веса и выражая каждый полученный таким путём атомный фактор в долях от их общей суммы, принятой за 100. Для кислорода подобный пересчёт даёт цифру 52,3. Таким образом, более половины всех составляющих земную кору атомов приходится на долю кислорода.
Древнейшая атмосфера Земли, по-видимому, не содержала свободного кислорода. Можно предполагать, что первичное его появление было обусловлено происходящим под действием ультрафиолетовых лучей Солнца разложением молекул водяного пара по общей схеме:
2 Н2О = 2 Н2 + О2.
Возникавший таким путём водород уходил вверх, а главная масса кислорода расходовалась на взаимодействие со способными окисляться веществами. Быстрое обогащение атмосферы кислородом началось, вероятно, лишь после появления на Земле растительности.
Кислород был открыт в 1774 г. Хотя вблизи земной поверхности атмосфера содержит его в виде молекул (О2), выше 100 км основной формой существования этого элемента становится атомарная. Распад молекул О2 на атомы осуществляется под воздействием ультрафиолетового излучения Солнца.
Соединение отдельных атомов кислорода в молекулы О2 сопровождается значительным выделением энергии (250 кДж/моль атомов). Есть предположение, что это может быть использовано для обеспечения полётов на больших высотах.
Свободный кислород состоит из двухатомных молекул. Под обычным давлением он сжижается при -183 °С и затвердевает при -219 °С. В газообразном состоянии кислород бесцветен, а в жидком и твёрдом имеет бледно-синюю окраску.
Критическая температура кислорода равна -118 °С, критическое давление 50 атм. Жидкий кислород имеет плотность 1,14 г/см3 (при температуре кипения) и характеризуется теплотой испарения 7 кДж/моль. Плотность твёрдого кислорода (при температуре плавления) равна 1,27 г/см3, а его теплота плавления 0,5 кДж/моль. Для твёрдого кислорода характерны кристаллы трёх различных типов, причём каждый из них устойчив в определённых пределах температур: ниже -249 °С, от -249 до -229 °С, и от -229 °С до температуры плавления. Пограничные значения температур между такими областями устойчивости (в данном случае -249 и -229 °С) носят названиеточек перехода.
Лабораторное получение кислорода основано на разложении богатых им, но сравнительно непрочных веществ. Обычно применяется хлорат калия (“бертолетова соль”), распадающийся при нагревании на хлорид калия и кислород:
2 KClO3 = 2 KCl + 3 O2.
Эта реакция интересна тем. что она значительно ускоряется и идёт при более низких температурах, если к KClO3 предварительно добавить немного двуокиси марганца (MnO2), количество которой после окончания процесса остаётся неизменным. Подобные двуокиси марганцавещества, ускоряющие реакции, но в результате их сами остающиеся химически неизменными, называются катализаторами. Каталитическая активность веществ специфична, т.е. какое-либо из них, служащее хорошим катализатором для одной реакции, нередко оказывается совершенно недеятельным при другой. Вместе с тем для реакции, катализируемой каким-либо одним веществом, можно обычно подобрать ещё ряд катализаторов. Так, при разложении KClO3 вместо MnO2 можно применить оксид железа (III) (Fe2O3), оксид хрома (III) (Cr2O3) и т.д.
Для получения медленного и равномерного тока кислорода вместо MnO2 к KClO3 примешивают измельчённую поваренную соль. Однако в этом случае нагревание должно быть более сильным. При точных работах следует иметь в виду, что полученный путём разложения КCIO3 кислород обычно содержит следы хлора.
Кислород может быть получен в лаборатории также рядом других методов, из которых наиболее удобны следующие: а) слабое прокаливание КМnO4; б) приливание по каплям раствора КМnO4 к подкисленному серной кислотой раствору Н2О2; в) действие воды в присутствии солей кобальта на пероксид натрия; г) действием разбавленной азотной кислоты на смесь равных весовых частей ВаО2 и РbO2; д) разложение воды, содержащей Н2SO4 или NaOH, постоянным электрическим током (одновременно образуется также водород). Для получения особо чистого кислорода (содержащего только примесь водяного пара) электролизу подвергают освобождённый кипячением от растворимых газов воздуха сернокислый раствор К2СrO4. Ежегодная мировая добыча кислорода исчисляется миллионами тонн.
В полевых условиях для получения кислорода удобно пользоваться тесной смесью 100 вес. ч. КCIO3 c 13 вес. ч. MnO2 и небольшим количеством угольной пыли. Смесь эта — оксигенит — начинает выделять кислород при её поджигании. Очистка от СО2 может быть осуществлена пропусканием выделяющегося газа сквозь сосуд с влажной гашеной известью.
Основным источником промышленного получения кислорода является жидкий воздух. Выделяемый из него кислород содержит обычно лишь незначительные количества примесей азота и тяжелых инертных газов. Для получения особо чистого кислорода пользуются иногда разложением воды электрическим током.
В 100 объёмах воды растворяется про 0 °С около пяти объёмов кислорода, при 20 °С — около трёх. Воды гидросферы содержат 1,5•1013 т растворённого кислорода. Растворимость его в воде имеет громадное значение для жизни, так как служит источником энергии живых организмов процесс дыхания осуществляется с участием растворённого кислорода.
Химическая сущность дыхания состоит в соединении углерода и водорода органических веществ с кислородом воздуха. Как у животных, так и у растений оно происходит в химическом смысле одинаково. Однако у растений параллельно протекает процесс питания: под действием солнечных лучей организм растений синтезирует необходимые ему органические вещества из углекислого газа и воды, причём свободный кислород возвращается в атмосферу. Общее его количество, выделяемое растениями в процессе дыхания, примерно в шесть раз больше потребляемого ими при дыхании.
Дыханию живых организмов аналогичны в химическом отношении протекающие повсюду разнообразные процессы окисления. В узком смысле слова под окислением понимается соединение вещества с кислородом. Так как последний является одним из самых активных химических элементов, он более или менее энергично реагирует почти со всеми остальными. Если окисление протекает с большим выделением тепла и света, то его называют горением. Медленно протекающие процессы окисления в зависимости от характера окисляющегося вещества называют ржавлением (для железа), тлением (для органических остатков) или чаще всего просто окислением.
Окислительные процессы протекают гораздо энергичнее в чистом кислороде, чем на воздухе. Например, тлеющая лучинка вспыхивает и ярко горит в кислороде, Такой же эффект из всех бесцветных газов даёт только гемиоксид азота, почти не встречающийся на практике. Поэтому проба на тлеющую лучинку часто служит для доказательства того, что испытываемый газ является именно кислородом.
Кислород широко применяется для получения высоких температур, которые достигаются путём сжигания различных горючих газов (водорода, светильного газа и т.д.) в смеси не с воздухом, а с чистым кислородом. Особенно распространено применение кислорода в смеси с ацетиленом (температура пламени около 3000 °С) для сварки и резки металлов. В медицине вдыхание кислорода иногда назначается при некоторых отравлениях, заболеваниях лёгких и др. Очень большое практическое значение имеет использование кислорода (чаще — обогащенного им воздуха) для интенсификации ряда важнейших производственных процессов металлургической и химической промышленности.
Кислород держат в голубых баллонах с чёрной надписью “Кислород”. Большие его количества хранят и перевозят в жидком состоянии. Для этого служат специальные ёмкости (“танки”) с хорошей теплоизоляцией. Исправный танк на 1 т теряет за час не более 4 кг кислорода (путём испарения сквозь отверстие в верхней части). Жидкий кислород применяется для заправки ракет.
Используемое в ракетах реактивное вещество обычно слагается из горючего вещества и окислителя. Оно должно одновременно удовлетворять ряду условий (скорость сгорания, теплотворная способность, температура пламени, характер продуктов сгорания и др.), далеко не всегда совместимых друг с другом. Важной числовой характеристикой такого топлива является его удельный импульс (удельная тяга). Чем он больше, чем меньший расход топлива требуется для получения заданной тяги. Удельный импульс определяется как отношение развиваемой тяги (кГ) к секундному расходу топлива (кГ/с) и обычно не превышает 300 с. Например, удельный импульс часто применяемой в небольших ракетах смеси спирта с кислородом (при наиболее принятых условиях сопоставления — давлении около 20 атм в камере сгорания) составляет примерно 250 с (а смеси керосина с кислородом — примерно 300 с).
В результате разнообразных процессов окисления кислород постоянно переходит из свободного состояния в связанное. Однако количество свободного кислорода остаётся практически неизменным, так как убыть его компенсируется жизнедеятельностью растений.
Озон.
В 1840 году было получено газообразное вещество, состоящее из молекул О3 и сильно отличающееся по свойствам от обычного кислорода (О2). Новый газ, обладающий характерным запахом, назвали озоном (по-гречески — “пахучий”).
Подобно обычному кислороду, озон представляет собой простое вещество. Он является аллотропной модификацией кислорода.
Газообразный озон голубоватого цвета, в жидком состоянии, в твёрдом — почти чёрным. Температура плавления озона -192 °С, температура кипения -112 °С. Во всех агрегатных состояниях озон способен взрываться от удара. Растворимость его в воде гораздо больше, чем кислорода.
При нормальном давлении озона 100 объёмов воды растворяют при обычных температурах около 45 объёмов этого газа. Ещё лучшим его растворителем является четырёххлористый углерод, один объём которого в тех же условиях поглощает около трёх объёмов озона. Такой раствор имеет красный цвет.
У земной поверхности озон образуется главным образом при грозовых разрядах и окислении некоторых органических веществ. В связи с этим заметные его количества содержатся в воздухе хвойных лесов, где окислению подвергается древесная смола, и на берегу моря, где окислению подвергаются выброшенные прибоем водоросли. Очень небольшое содержание озона в воздухе благотворно действует на организм человека, особенно при болезнях дыхательных путей.
Среднее содержание озона в воздухе у земной поверхности составляет обычно от 0,01 до 0,06 мг/м3. Общее его содержание в атмосфере соответствует слою газа толщиной приблизительно в 3 мм (при нормальном давлении). Основная масса озона сосредоточена в высоких слоях воздуха (10-30 км), где он образуется из кислорода под действием ультрафиолетовых лучей Солнца с длиной волны до 185 нм. Более длинные волны (200-320 нм с максимумом действия при 255 нм) вызывают, наоборот, распад озона. Поэтому, в атмосфере существует подвижное между процессами образования и распада озона, на поддержание которого затрачивается около 5% всей идущей к Земле солнечной энергии. Поглощение озоном коротковолнового излучения Солнца имеет очень большое биологическое значение: если бы эти “жёсткие” лучи свободно достигали земной поверхности, они быстро убили бы всю жизнь на ней.
Запах озона становится заметным при концентрации его более 1:109 по объёму. Продолжительное пребывание в атмосфере с содержанием озона порядка 1:106 вызывает раздражительность, чувство усталости и головную боль. При более высоких концентрациях к этим симптомам добавляется тошнота, кровотечение из носа и воспаление глаз. В производственных условиях озон может образовываться всюду, где происходят электрические разряды или действует коротковолновое излучение. Повышенное его содержание часто обнаруживается, например, в рентгеновских кабинетах. Максимально допустимой концентрацией озона в закрытых помещениях считается 0,1 мг/м3.
Получают озон чаще всего действием на газообразный кислород тихого разряда (электрического разряда без свечения и искр). Применяемый для этого прибор — озонатор. Тихий разряд происходит в пространстве между стенками внутреннего и внешнего стеклянных сосудов. Выходящий из озонатора кислород содержит несколько процентов озона. Его образование сопровождается уменьшением объёма, так как по реакции: 3 О2Ы 2 О3 из трёх объёмов кислорода получается два объёма озона.
Более или менее значительный процент озона содержится в кислороде, образующимся при распаде различных пероксидных соединений. Небольшие количества озона можно получать нагреванием (в пробирке) персульфата аммония с концентрированной азотной кислотой или действием концентрированной серной кислоты на пероксид бария. С хорошим выходом —более 20 вес.% — озон может быть получен в больших количествах электролизом концентрированных (40 вес.%) водных растворов хлорной кислоты при низких температурах (ниже -50 °С) и уменьшенном давлении (0,1 атм).
Озон сравнительно легко самопроизвольно переходит в кислород, что сопровождается значительным выделением энергии. Следовательно, образование озона связано с поглощением такого же количества энергии. Это вытекает из общего принципа термохимии, согласно которому при образовании любого соединения поглощается (выделяется) точно такое же количество энергии, какое выделяется (поглощается) при его распаде на исходные вещества.
Это по существу частный случай закона сохранения и превращения энергии: энергия не возникает из нечего и не исчезает бесследно, но отдельные её виды могут переходить друг в друга по строго определённым эквивалентным соотношениям.
Термохимия изучает энергетические изменения при химических превращениях. В зависимости от характера процесса и условий его протекания энергия может выделяться или поглощаться в различных формах. Ввиду взаимной эквивалентности отдельных видов энергии все они могут быть выражены в тепловых единицах.
Реакции, протекающие с выделением теплоты, называются экзотермическими, протекающие с его поглощением — эндотермическими. Выделение или поглощённое количество энергии может быть указано в уравнении реакции, причём он относится к тому числу молей вещества, которое входит в уравнение. Так, для распада и образования озона имеет:
экзотермическая реакцияЮ
2 О3 = 3 О2 + 284 кДж
Ьэндотермическая реакция
Уравнение показывает, что при распаде (образовании) двух молей озона (96 г) выделяется (поглощается) 284 кДж.
“Вопрос о количестве теплоты, выделяемой или поглощаемой при химических реакциях, очень сложен, так как рядом с химическим процессом имеют место и физические явления, также могущие влиять на термическую сторону дела”, — писал Д.И. Менделеев в 1875 г. В частности, на общее выделение или поглощение энергии при той или иной химической реакции более или менее существенное влияние оказывает переход реагирующих веществ из одного агрегатного состояния в другое, как все подобные переходы связаны с выделением или поглощением энергии.
В термохимии агрегатное состояние исходных веществ и получающихся продуктов условно обозначаются, заключая формулы твёрдых при условиях протекания реакции веществ в квадратные скобки, жидких — в фигурные (или оставляя их без скобок) и газообразных — в круглые. Другой часто применяемый способ обозначения агрегатных состояний использует начальные буквы их названий — (г), (ж) и (т) в виде индексов при формулах.
Термохимические уравнения часто относят к одной моль получившегося вещества. В соответствии с этим реакция распада озона записывает следующим образом:
2/3 (О3) = (О2) + 95 кДж или 2/3 О3(г) = О2(г) + 95 кДж.
При отсутствии указаний относительно агрегатных состояний входящих в уравнение веществ подразумевается, что они находятся в том виде, который соответствует условиям протекания реакции, а если эти условия не оговорены, то обычным условиям (комнатная температура, атмосферное давление).
Химические процессы проводятся обычно под неизменным (чаще всего — атмосферным) давлением, но при различных температурах, причём изменение температуры влияет на тепловой эффект. Например, для реакции по уравнению
2 (SO2) + (O2) = 2 (SO3) + Q имеем:
Температура, °С | 25 | 400 | 500 | 600 | 700 |
| Q, кДж | 196 | 190 | 188 | 186 | 184 |
Подобным же образом теплота синтеза аммиака по уравнению:
(N2) + 3 (H2) = 2 (NH3) + Q,
равная 92 кДж при 25 °С, составляет 106 кДж при 500 °С и 110 кДж при 660 °С. Так как научная литература по международному соглашению (в целях сопоставимости) обычно приводит данные, отнесённые к 25 °С, ими приходится пользоваться для оценки тепловых эффектов реакций, протекающих и при других температурах. Приведённые примеры показывают, что привносимые этим ошибки, как правило, невелики.
Следует также отметить различие знаков тепловых эффектов, принятых в термохимии и термодинамике. Термохимия рассматривает энергетику процессов с точки зрения их наблюдателя, т. е. положительным знаком отмечает выделение тепла при реакциях. Напротив, термодинамика рассматривает процессы с точки зрения увеличения или уменьшения запаса энергии в самих веществах и положительным знаком отмечает поглощение тепла. Поэтому при пользовании справочниками необходимо прежде всего установить проводимую в них систему обозначений. В настоящей книге принята термохимическая система.
Сами соединения, образующиеся с выделением энергии, называются экзотермичными, а образующиеся с поглощением энергии — эндотермичными. Подобные озону эндотермичные вещества всегда имеют склонность к распаду (и тем большую, чем более они эндотермичны). Все они, следовательно, более или менее неустойчивы. Однако многие из них всё же можно сохранить, так как при обычных условиях разложение практически не идёт. В частности, это относится к озону, смешанному с избытком кислорода. Вместе с тем чистый озон чрезвычайно взрывчат, и поэтому работы с ним весьма опасны.
Распад молекулы озона инициируется, по-видимому, её столкновением с какой-либо другой частицей (Х). Действительный ход превращения озона в обычный кислород хорошо описывается следующими уравнениями:
О3 + Х = Х + О2 + О
и затем О + О3 = 2 О2.
Почленное сложение этих двух реакций (с сокращением одинаковых членов) приводит к: 2 О3 = 3 О2.
Взрывоопасность озона резко уменьшается при полном исключении возможности его соприкосновения даже со следами способных окисляться веществ. Эффективным путём повышения стабильности (устойчивости) озона является предварительное пропускание исходного молекулярного кислорода сквозь слой нагретого до 700 °С оксида меди. Газообразные смеси озона с кислородом не взрывообразны лишь при содержании в них более 80 объёмн. % кислорода.
Молекула О3 легко отдаёт один атом кислорода. Поэтому озон является сильным окислителем. Под его действием почти все металлы (кроме Аи, Рt и Ir) превращаются в оксиды, сернистые соединения окисляются в сернокислые, аммиак — в азотистую и азотную кислоты и т.д. Резина очень быстро разрушается озоном, а многие другие органические вещества (например, спирт) при соприкосновении с ним воспламеняются. Эта исключительно высокая окислительная активность озона и является его наиболее характерным химическим свойством.
После некоторого поверхностного окисления довольно хорошо противостоят действию озона Сu, Ni и Sn. Не разрушается озоном также сплав железа (не содержащий углерода) с 25% хрома.
Практическое применение озона основано на его сильном окисляющем и стерилизующем действии. Под действием озона погибают не только бактерии, но и грибковые образования и вирусы. Озонированным воздухом пользуются для дезинфекции помещений (холодильных складов и др.), устранения неприятных запахов (в курительных комнатах и т.д.), стерилизацией питьевой воды, кондиционирования воздуха и проведением других окислительных процессов. Сжигание горючих веществ а атмосфере озона создаёт возможность резкого ускорения сгорания веществ в атмосфере озона создаёт возможность резкого ускорения сгорания и получения более высоких температур, чем при сжигании тех же веществ в кислороде. Поэтому озон представляет большой интерес для реактивной техники.
ОСНОВНЫЕ КЛАССЫ НЕОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Оксиды.
Оксидами называютсясоединения элементов с кислородом, в которых кислород соединяется только с атомами других элементов, например K–O–K, Са=О, О=Si=О. Соединения элементов с кислородом, в которых осуществляется связь между двумя атомами кислорода, называются перекисями (например, K–O–O–K). Почти все элементы образуют соединения с кислородом. В одних случаях оксиды образуются при непосредственном взаимодействии простых веществ с кислородом, в других — их получают косвенным путём.
Многие оксиды способны присоединять воду, образуя основания или кислоты. Все оксиды подразделяют на солеобразующие и несолеобразующие.
Неслеобразующих оксидов немного. К ним, в частности, принадлежат гемиоксид азота N2O, монооксид азота NO, монооксид углерода СО и некоторые другие. Их иногда называют индифферентными (безразличными), однако это название нельзя признать удачным, так как перечисленные оксиды вовсе не индифферентны и легко вступают в различные химические реакции.
Солеобразующие оксиды подразделяются на основные, кислотные и амфотерные.
Основные оксиды. Основными называются оксиды, образующие соли при взаимодействии с кислотами или кислотными оксидами. К основным оксидам принадлежат, например, Li2O, Na2O, K2O, Ag2O, MgO, CaO, SrO, BaO, HgO, MnO, FeO, NiO. Основными могут быть только оксиды металлов. Но не все оксиды металлов являются основными: многие из них принадлежат к амфотерным или кислотным. Ниже приведены примеры образования солей основными оксидами:
MgO + 2 HCl = MgCl2 + H2O
CaO + CO2 = CaCO3.
Кислотные оксиды. Кислотными называются оксиды, образующие соли при взаимодействии с основаниями или основными оксидами:
CO2 + 2 KOH = K2CO3 + H2O
SrO + SO3 = SrSO4.
Кислотные оксиды называют также ангидридами кислот. Многие из них соединяются с водой, образуя кислоты. Кислотными являются, например, оксиды типичных неметаллов: B2O3, N2O3, P2O3, P2O5, SO2, CO2, NO2, SiO2.
Кислотный характер имеют также оксиды некоторых металлов в степени окисления пять и выше:
V2O5 + Ca(OH)2 = CaVO3 + H2O
CrO3 + 2 NaOH = Na2CrO4 + H2O
Mn2O7 + 2 KOH = 2 KMnO4 + H2O.
Амфотерные оксиды. Амфотерными называются оксиды металлов, образующие соли при взаимодействии как с кислотами (кислотными оксидами), так и с основаниями (основными оксидами).
К амфотерным оксидам относятся ZnO, SnO, PbO, Cr2O3, Al2O3, MnO2, SnO2, PbO2, Fe2O3 и др. Амфотерный характер оксида алюминия, например, проявляется во взаимодействии его как с соляной кислотой, так и с гидроксидом калия:
2 Al2O3 + 6 HCl = 2 AlCl3 + 3 H2O
Al2O3 + 2 KOH = 2 KAlO2 + H2O.
И в одном и в другом случае оксид алюминия образует соль. В первой реакции оксид алюминия ведёт себя как основной оксид, и полученную соль можно рассматривать как продукт замещения водорода соляной кислоты алюминием. Во второй реакции оксид алюминия выступает как кислотный: он образует соль, в которой алюминий входит в состав кислотного остатка
Следовательно, амфотерным оксидам присущи свойства как основных, так и кислотных свойств. У различных амфотерных оксидов эта двойственность может быть выражена в различной степени. Например, оксид цинка ZnO одинаково легко растворяется и в кислотах, и в щелочах, т. е. у этого оксида основная и кислотная функции выражены примерно в равной мере. Fe2O3 обладает преимущественно основными свойствами и кислотные свойства проявляет, только взаимодействуя со щелочами при высоких температурах. У амфотерного диоксида олова SnO2 преобладают кислотные свойства.
Номенклатура оксидов. Если элемент образует с кислородом только одно соединение, его называют оксидом независимо от состава. Например, Na2O — оксид натрия, CaO — оксид кальция, Al2O3 — оксид алюминия и т. п.
Если же элемент образует несколько оксидов, то их названия даются с учётом состава оксида. Например:
N2О — гемиоксид азота, NO — монооксид азота, N2О3 — сесквиоксид азота, NO2 — диоксид азота, N2O5 — сесквиоксид азота, SО3 — триоксид серы, Fe3О4 — оксид железа (II, III). Если элемент обладает переменной валентностью, то после слов оксид элемента в круглых скобках римскими цифрами указывают его валентность, например, оксид фосфора (V), оксид железа (III) и т.д.
Способы получения оксидов.
1. Взаимодействие простых веществ с кислородом. Многие простые вещества при нагревании на воздухе или в кислороде сгорают, образуя соответствующие оксиды:
2 Мо + 3 О2 = 2 МоО3
4 Р + 5 О2 = 2 Р2О5.
2. Разложение оснований. Некоторые основания при нагревании теряют воду, превращаясь в оксиды металлов:
Ва(ОН)2 = ВаО + Н2О
2AI(OH)3 = AI2O3 + 3 H2O.
Реакции протекают с различной степенью лёгкости. Так, образование оксида ртути и оксида серебра из гидроксидов этих металлов происходит уже при комнатной температуре:
Hg(OH)2 = HgO + H2O
2 AgOH = Ag2O + H2O.
Напротив, гидроксид натрия можно перегнать при 1390 °С, без разложения.
3. Разложение кислот. Кислородсодержащие кислоты при нагревании теряют воду, превращаясь в кислотные оксиды:
Н4SiO4 = SiO2 + 2 H2O
4 HNO3 = 4 NO2 + 2 H2O + O2.
Иногда можно достичь удаление воды из кислородсодержащих кислот действием на них водоотнимающих средств:
2 HCIO4 + P2O5 = 2 HPO3 + CI2O7
2 HNO3 + P2O5 = 2 HPO3 + N2O5
Некоторые кислоты самопроизвольно теряют воду даже при низких температурах:
H2CO3 = H2O + CO2
H2SO3 = H2O + SO2.
4. Разложение солей. Подавляющее большинство солей кислородсодержащих кислот при нагревании разлагается на оксид металла и кислотный оксид:
СаСО3 = СаО + СО2
Fe2(SO4)3 = Fe2O3 + 3 SO3
2 Pb(NO3)2 = 2 PbO + 4 NO2 + O2.
Если оксид металла термически неустойчив, то вместо оксида образуется свободный металл:
2 Ag2CO3 = 4 Ag + 2 CO2 + O2
Hg(NO3)2 = Hg + 2 NO2 + O2.
Cоли щелочных металлов отличаются высокой термической устойчивостью. Если они при нагревании всё же разлагаются, то оксиды при этом, как правило, не образуются:
2 KNO3 = 2 KNO2 + O2
2 KCIO3 = 2 KCI + 3 O2.
5. Разложение оксидов. Если элемент имеет переменную валентность, то его оксид с меньшим содержанием кислорода можно получить нагреванием оксида, в котором элемент проявляет более высокую степень окисления:
2 SO3 = 2 SO2 + O2
2 N2O5 = 2 NO2 + O2
4 CrO3 = 2Cr2O3 + 3 O2.
И наоборот, высшие оксиды иногда удаётся получить окислением низших:
2 СО + О2 = 2 СО2
6 PbO + O2 = 2 Pb3O4
P2O3 + O2 = P2O5.
6. Вытеснение одних оксидов другими. Эта реакция может быть применена для получения более летучих оксидов вытеснением их менее летучими:
СаСО3 + SiO2 = CaSiO3 + CO2
CoSO4 + B2O3 = Co(BO2)2 + SO3.
Реакции протекают при высокой температуре и основаны на том, что сесквиоксид бора и диоксид кремния нелетучи и при нагревании вытесняют более летучие: диоксид углерода и триоксид серы.
7. Взаимодействие кислот, обладающих окислительными свойствами, с металлами и неметаллами. Азотная и концентрированная серная кислоты при действии восстановителей образуют оксиды, в которых азот и сера проявляют более низкую степень окисления, чем в исходных кислотах.
Cu + 4 HNO3 = Cu(NO3)2 + 2 NO2 + 2 H2O
C + 2 H2SO4 = CO2 + 2 SO2 + 2 Н2O.
Пероксиды. Некоторые соединения металлов с кислородом по химическим свойствам существенно отличаются от обычных оксидов. Так, соединения Na2O2, BaO2, ZnO2 состоят из металла и кислорода, но являются не оксидами, а солями пероксида водорода и поэтому называются пероксидами. У пероксидов связанные друг с другом атомы кислорода образуют не очень прочную пероксидную группу -О-О-. Поэтому при действии кислот на пероксиды металлов наряду с солями образуется кислород:
О—Na
2 Ѕ + 2 H2SO4 = 2 Na2SO4 + 2 H2O + O2
О—Na
2 BaO2 + 4 HNO3 = 2 Ba(NO3)2 + 2 H2O + O2.
Смешанные оксиды. Соединения Pb2O3, Mn3O4, Fe3O4 иногда называют двойными или смешанными оксидами. Их можно также рассматривать как соли: Pb2O3є PbPbO3 — плюмбат свинца (соль свинцовой кислоты H2PbO3); Mn3O4є Mn2MnO4 — манганит марганца (соль H4MnO4); Fe3O4є Fe(FeO2)2 — феррит железа (II) (соль НFeO2). Следовательно, в состав молекулы смешанного оксида входят атомы одного элемента в различных степенях окисления.
Соединения оксидов с водой называют гидратами оксидов. Присоединение оксидом воды не приводит к коренному изменению его химического характера, поэтому гидраты основных оксидов проявляют основные свойства, гидраты амфотерных оксидов — амфотерные, а гидраты кислотных оксидов имеют кислотные свойства.
Основания (гидроксиды).
Основаниями называют гидраты основных оксидов. Общая формула оснований — М(ОН)n. Количество гидроксильных групп в молекуле основания определяет его кислотность. Например, NaОН — однокислотное основание, AI(OH)3 — трёхкислотное основание.
Большинство оснований нерастворимо в воде. Растворимы гидроксиды щелочных и щелочноземельных металлов и гидроксид аммония. В водных растворах такие основания диссоциируют на положительно заряженные ионы металла и отрицательно заряженные ионы гидроксила. Многокислотные основания диссоциируют ступенчато:
Са(ОН)2Ы СаОН+ + ОН-
СаОН+Ы Са2+ + ОН-.
Основания растворимые в воде и хорошо диссоциированные, называются щелочами. Примеры щелочей: LiOH, NaOH, KOH, RbOH, CsOH, Sr(OH)2, Ba(OH)2, Ca(OH)2.
Раствор аммиака в воде проявляет свойства слабого основания, так как на ионы распадается незначительное количество молекул гидроксида аммония NH4OH.
Основания, как и основные оксиды, взаимодействуют с кислотами или кислотными оксидами, образуя соли:
Ni(OH)2 + H2SO4 = NiSO4 + 2 H2O
Ca(OH)2 + CO2 = CaCO3Ї + H2O.
Способы получения оснований.
1. Взаимодействие активных металлов с водой. Щелочные и щелочноземельные металлы уже при комнатной температуре разлагают воду, образуя основания:
2 K + 2 H2O = 2 KOH + H2
Ca + 2 H2O = Ca(OH)2 + H2.
2. Непосредственное соединения основных оксидов с водой. Подавляющее большинство основных оксидов непосредственно с водой не соединяется. Только оксиды щелочных и щелочноземельных металлов, присоединяя воду, образуют основания:
Li2O + H2O = 2 LiOH
BaO + H2O = Ba(OH)2.
3.Взаимодействие солей со щелочами. Этот метод применяют главным образом для получения нерастворимых в воде оснований:
CuSO4 + 2 KOH = Cu(OH)2Ї + K2SO4
FeCI3 + 3 NaOH = Fe(OH)3Ї + 3 NaCI.
Получение растворимых оснований по этому методу возможно в случае, когда в результате реакции образуется нерастворимая соль:
K2CO3 + Ca(OH)2 = CaCO3Ї + 2 KOH
Na2SO4 + Ba(OH)2 = BaSO4Ї + 2 NaOH.
4. Электролиз растворов солей. Этот метод применяется при получении щелочей в технике, для чего пропускают постоянный электрический ток через водные растворы солей натрия или калия. Например, при электролизе водного раствора хлорида натрия на катоде выделяется водород, на аноде — хлор, а в растворе накапливается гидроксид натрия. Упаривая такой раствор, получают кристаллический гидроксид натрия. Процессы, происходящие при электролизе раствора хлорида натрия, можно представить следующей схемой:
NaCI Ы Na+ + CI-
H2O Ы H+ + OH-
На катоде: На аноде:
Н+ + е- = Н СI-- e- = CI
2 H = H2 2 CI = CI2
В растворе в катодном пространстве остаётся NaOH.
Кислоты.
Кислотами называются соединения, которые при электролитической диссоциации образуют ионы водорода и других ионов не дают. В водных растворах кислоты диссоциируют на ионы водорода и кислотный остаток. Количество атомов водорода, способных замещаться металлами с образованием солей, определяет основность кислоты. Различают кислоты одноосновные (HCI, HNO3), двухосновные (H2SO4, H2S), трёхосновные (Н3РО4), шестиосновные (H6V10O28).
В некоторых кислотах не все атомы водорода способны замещаться металлами. Например, молекула уксусной кислоты СН3СООН содержит четыре атома водорода, однако замещаться металлами способен лишь атом водорода карбоксильной группы СООН, поэтому уксусная кислота является одноосновной. Фосфористая кислота Н3РО3 — двухосновная, фосфорноватистая Н3РО2 — одноосновная.
По химическому составу различают кислоты бескислородные и кислородсодержащие. Примерами бескислородных кислот могут служить плавиковая (НF), соляная (НСI), бромводородная (НВr), иодоводородная (НI), циановодородная (синильная НСN), родановодородная (НСNS), сероводородная (H2S).
Кислородсодержащие кислоты представляют собой гидраты кислотных оксидов. Большинство кислотных оксидов образует кислоты в результате непосредственного присоединения воды. Молекулы некоторых ангидридов при разных условиях могут присоединять различные количества молекул воды, образуя соединение с бульшим содержанием воды — ортокислоту — и соединение с меньшим содержанием воды — метакислоту. Например:
Р2О5 + Н2О = 2 НРО3 — метафосфорная кислота
Р2О5 + 3 Н2О = 2 Н3РО4 — ортофосфорная кислота
В2О3 + Н2О = 2 НВО2 — метаборная кислота
В2О3 + 3 Н2О = 2 Н3ВО3 — ортоборная кислота
Диоксид азота NO2 при взаимодействии с водой даёт две кислоты — азотную и азотистую:
2 NO2 + H2O = HNO2 + HNO3.
Аналогичным образом ведёт себя диоксид хлора, образующий с водой хлорноватую и хлористую кислоты:
2 CIO2 + H2O = HCIO3 + HCIO2.
Кислотные оксиды, образованные при взаимодействии с содой две кислоты, называются смешанными ангидридами. При взаимодействии их с основаниями, естественно, образуются две соли.
Многоосновные кислоты в растворах диссоциируют ступенчато:
Н3РО4Ы Н2РО4- + Н+
Н2РО4-Ы НРО42- + Н+
НРО42-Ы РО43- + Н+.
Номенклатура кислот. Если элемент, обладающий переменной валентностью, образует несколько кислот, то для их различия в названии используют разные суффиксы. Так, если элемент образует две кислоты, то для обозначения той из них, в которой кислотообразующий элемент имеет более низкую валентность, используют суффикс -ист: H2SO4 — серная кислота, H2SO3 — cернистая, НNO3 — азотная кислота, HNO2 — азотистая кислота.
Когда элемент образует более двух кислородсодержащих кислот, для их обозначения употребляют суффиксы “оват”, “ист” и “оватист”:
HCIO4 — хлорная H3PO4 — фосфорная
HCIO3 — хлорноватая H2PO3 (H4P2O6)— фосфорноватая
HCIO2 — хлористая H3PO3 — фосфористая
HCIO — хлорноватистая H3PO2 — фосфорноватистая.
Для обозначения кислот, получаемых частичным обезвоживанием ортокислот, пользуются приставкой пиро-:
2 Н3РО4 = Н2О + Н4Р2О7 — пирофосфорная кислота.
Способы получения кислот.
1. Взаимодействие ангидридов кислот с водой. Большинство ангидридов способно непосредственно присоединять воду, образуя соответствующие кислоты:
SO3 + H2O = H2SO4
P2O5 + 3 H2O = 2 H3PO4
N2O5 + H2O = 2 HNO3
2. Взаимодействие солей с кислотами. Это наиболее распространённый способ:
2 NaCI + H2SO4 = Na2SO4 + 2 HCI
NaNO3 + HPO3 = NaPO3 + HNO3.
При получении кислот этим способом исходная соль должна быть достаточно растворимой, а взятая для реакции кислота — более сильной или менее летучей, чем получаемая. Серная кислота является сильной и нелетучей, поэтому ею часто пользуются для получения других кислот. Если получаемая кислота обладает восстановительными свойствами, то вместо серной кислоты для реакции берут фосфорную кислоту.
3. Окисление некоторых простых веществ. Кислоты получаются при действии на некоторые неметаллы сильных окислителей:
I2 + 5 Cl2 + 6 H2O = 2 HIO3 + 10 HCl
3 P + 5 HNO3 + 2 H2O = 3 H3PO4 + 5 NO.
4. Взаимодействие неметаллов с водородом. Некоторые бескислородные кислоты можно получить непосредственным соединением неметалла с водородом:
H2 + I2 = 2 HI
H2 + Cl2 = 2 HCl.
Водные растворы полученных соединений являются кислотами.
Амфотерные гидроксиды.
Гидраты амфотерных оксидов, как и сами оксиды, обладают амфотерными свойствами. Эти соединения весьма мало растворимы в воде. Если записать формулу амфотерного гидроксида в общем виде как М(ОН)x, то диссоциацию гидроксида в растворе по основному и кислотному типам можно представить схемой:
М(ОН)хЫ Мх+ + х ОН-
М(ОН)хЫ х Н+ + МОхх-.
Поскольку амфотерные гидроксиды диссоциируют по основному и кислотному типам одновременно, этот процесс можно записать следующим образом:
Мх+ + х ОН-Ы М(ОН)хє НхМОхЫ х Н+ + МОхх-.
В насыщенном растворе амфотерного гидроксида ионы Мх+, ОН-, Н+ и Мохх- находятся в состоянии равновесия. Поэтому амфотерные гидроксиды взаимодействуют как с кислотами, так и с основаниями, образуя соли.
При взаимодействии с кислотами амфотерные гидроксиды проявляют основные свойства:
Zn(OH)2 + H2SO4 = ZnSO4 + 2 H2O
Al(OH)3 + 3 HCl = AlCl3 + 3 H2O.
Продукты реакций — сульфат цинка и хлорид алюминия — содержат металл в виде катиона.
Взаимодействуя со щелочами, гидраты амфотерных оксидов проявляют кислотные свойства:
H2ZnO2 + 2 KOH = K2ZnO2 + 2 H2O
H3AlO3 + 3 NaOH = Na3AlO3 + 3 H2O.
Образовавшиеся соли (цинкат калия и алюминат натрия) содержат соответственно цинк и алюминий в составе кислотного остатка.
Взаимодействие гидроксида алюминия с гидроксидом натрия может протекать и по другой схеме:
H3AlO3 + NaOH = NaAlO2 + 2 H2O.
Образовавшуюся соль называют метаалюминатам натрия в отличие от соли Na3AlO3, называемой ортоалюминатом натрия. Образование орто- или метасоединения определяется концентрацией щёлочи и условиями реакции: ортоалюминаты образуются в растворах, метаалюминаты — при сплавлении.
(Взаимодействие амфотерных гидроксидов со щелочами в растворах происходит по уравнениям:
Zn(OH)2 + 2 KOH = K2[Zn(OH)4]
Al(OH)3 + 3 NaOH = Na3[Al(OH)6] )
Гидраты амфотерных оксидов обычно получают взаимодействием солей со щёлочью, количество которой рассчитывают по уравнению реакции, например:
ZnSO4 + 2 NaOH = Zn(OH)2Ї + Na2SO4
Cr(NO3)3 + 3 KOH = Cr(OH)3Ї + 3 KNO3.
Соли.
Солью называют продукт замещения атомов водорода в кислоте на металл. Растворимые в воде соли диссоциируют на катион металла и анион — кислотный остаток. Соли подразделяют на средние, кислые и основные.
Средние соли являются продуктами полного замещения атомов водорода в кислоте на металл: MgSO4, Al2(SO4)3, Na3PO4. Атомы водорода в кислоте могут быть замещены группой атомов, играющей роль катиона. Например, вместо водорода может стоять аммонийная группа NH4+: NH4Cl, (NH4)2SO4, (NH4)3SO4.
Иногда атом металла в средней соли бывает связан с двумя различными кислотными остатками; такие соли называют смешанными. Примером смешанной соли может служить белильная известь, являющаяся кальциевой солью двух кислот — соляной и хлорноватистой: Cl-Ca-O-Cl.
Если атомы водорода многоосновной кислоты замещены двумя различными металлами, то такую соль называют двойной, например NaKCO3, Na2NH4PO4, KAI(SO4)2.
Двойные и смешанные соли как индивидуальные соединения известны только в кристаллическом состоянии, в растворах они полностью диссоциированы на ионы металлов и кислотные остатки.
Название средних солей производят от названий образовавших их кислот и металлов:
CuSO4 — сульфат меди, K2SO3 — сульфит калия, Na2CO3 — карбонат натрия, Mg(NO3)2 — нитрат магния, NaNO2 — нитрит натрия, NaCI — хлорид натрия NaCIO — гипохлорит натрия, NaCIO2 —хлорит натрия, КCIO3 — хлорат калия, NaCIO4 — перхлорат натрия.
Иногда при наименовании средних солей пользуются техническими названиями, например:
NaCI — поваренная соль, NaCO3 — сода кальцинированная,Na2CO3•10H2O — сода кристаллическая, K2CO3 — поташ, KNO3 — калийная селитра, KAI(SO4)2•12H2O — алюмокалиевые квасцы.
Кислые соли можно рассматривать, как продукты неполного замещения атомов водорода в кислоте на металл. Образование кислых солей характерно только для многоосновных кислот. Кислые соли состоят из металла, кислотного остатка, и водорода, способного замещаться металлами. В водных растворах кислые соли диссоциируют на отрицательно заряженные ионы кислотных остатков и положительно заряженные ионы двух видов — металла и водорода, например:
NaHSO4Ы Na+ + H+ + SO42-.
Кислые соли чаще всего образуются при избытке кислоты и могут быть переведены в средние соли действием оснований:
Са(HCO3)2 + Ca(OH)2 = 2 CaCO3 + 2 H2O
MgHPO4 + NH4OH = MgNH4PO4 + H2O.
Название кислых солей образуют из названий средних солей, добавляя приставку гидро-:
NaHCO3 — гидрокарбонат натрия; KHSO4 — гидросульфат калия.
При наименовании кислых солей многоосновных кислот указывают число ещё не замещённых атомов водорода:
NaH2PO4 — дигидрофосфат натрия; Na2HPO4 — гидрофосфат натрия.
Основные соли содержат помимо металла и кислотного остатка гидроксильные группы ОН-. Такие соли можно рассматривать как продукты неполного замещения гидроксильных групп основания кислотными остатками. Основные соли дают только многокислотные основания: AI(OH)2CI, Ni(OH)NO3, AI(OH)SO4, Cu2(OH)2CO3 и т. п. Все основные соли труднорастворимы в воде. Они обычно образуются при недостатке кислоты и могут быть переведены в средние соли действием кислот, например:
Ni(OH)NO3 + HNO3 = Ni(NO3)2 + H2O
Fe(OH)2CI + 2 HCI = FeCI3 + 2 H2O.
Названия основных солей чаще всего выводятся из названий средних, применяя приставку гидрокси-. Если гидроксильных групп в молекуле основной соли больше одной, то количество их указывают приставками ди-, три-, тетра- и т. д.:
Аl(OH)Cl2 — гидроксихлорид алюминия
Сr(OH)2NO3 — дигидроксинитрат хрома
Тi(OH)3CI — тригидроксихлорид титана
Основные соли при нагревании или со временем способны терять воду. Образующиеся при этом соли, которые, естественно, тоже имеют основной характер, называются оксисолями. Например:
2 Mg(OH)Cl = H2O + Mg2OCl2 — оксихлорид магния
2 Al(OH)SO4 = H2O + Al2O(SO4)2 —оксисульфат алюминия
Al2(OH)4SO4 = H2O + AI2O2SO4 — диоксисульфат алюминия
Оксисоли можно перевести в средние соли действием соответствующих кислот:
Zn2OCl2 + 2 HCl = 2 ZnCl2 + H2O.
Важнейшие способы получения солей.
1. Взаимодействие металла с кислотой. Образование солей при взаимодействии металла с кислотами может сопровождаться или не сопровождаться выделением водорода. Это зависит от активности металла, химических свойств кислоты и её концентрации.
Кислоты, не являющиеся окислителями, взаимодействуют лишь с металлами, находящимися в ряду напряжений левее водорода. В этих случаях образование солей сопровождается выделением водорода:
Zn + 2 HCl = ZnCl2 + H2
Mg + 2 CH3COOH = Mg(CH3COO)2 + H2.
Металлы, находящиеся в ряду напряжений правее водорода, с такими кислотами не взаимодействуют.
Кислоты, обладающие окислительными свойствами, вступают в реакцию как с активными, так и с малоактивными металлами без выделения водорода:
3 Mg + 8 HNO3 = 3 Mg(NO3)2 + 2 NO + 4 H2O
Cu + 4 HNO3 = Cu(NO3)2 + 2 NO2 + 2 H2O.
Характер взаимодействия с металлами серной кислоты существенно зависит от её концентрации. Разбавленная серная кислота не проявляет окислительных свойств и взаимодействует с активными металлами с выделением водорода:
Fe + H2SO4 = FeSO4 + H2.
Концентрированная серная кислота является окислителем и взаимодействует с металлами с образованием солей без выделения водорода:
Cu + 2 H2SO4 = CuSO4 + SO2 + 2 H2O
2. Взаимодействие основного оксида с кислотой:
СаО + 2 НCl = CaCI2 + H2O
FeO + H2SO4 = FeSO4 + H2O.
3. Взаимодействие основания с кислотой. Реакции этого типа имеют большое практическое значение и получили название реакции нейтрализации. Они всегда сопровождаются образованием воды.
Ва(ОН)2 + 2 НCl = BaCl2 + 2 H2O
2 NaOH + H2SO4 = Na2SO4 + 2 H2O.
4.Взаимодействие соли с кислотой. При реакциях этого типа образуется новая соль и новая кислота. Для осуществления этой реакции необходимо, чтобы взятая кислота была сильнее образующейся или менее летучей. Например:
СаСO3 + 2 HNO3 = Ca(NO3)2 + CO2 + H2O
2 NaCl + H2SO4 = Na2SO4 + 2 HCl.
Действием избытка кислоты на средние соли многоосновных кислот получают кислые соли:
Na2SO4 + H2SO4 = 2 NaHSO4
CaCO3 + CO2 + H2O = Ca(HCO3)2.
5. Взаимодействие основного оксида с кислотным:
СаО + SiO2 = CaSiO3
Ag2O + SO3 = Ag2SO4.
6. Взаимодействие основания с кислотным оксидом:
6 NaOH + P2O5 = 2 Na3PO4 + 3 H2O
2 KOH + CrO3 = K2CrO4 + H2O.
7. Взаимодействие соли с кислотным оксидом. Реакции этого типа происходят преимущественно при нагревании, поэтому вступающий в реакцию кислотный оксид должен быть менее летуч, чем образующийся после реакции:
СаСО3 + SiO2 = CaSiO3 + CO2
Cr2(SO4)3 + 3 B2O3 = 2 Cr(BO2)3 + 3 SO3.
8. Взаимодействие основания с солью. Этой реакцией часто пользуются в практике как для получения солей, так и для получения оснований, основных солей, для перевода кислых солей в средние:
Fe(NO3)3 + 3 NaOH = 3 NaNO3 + Fe(OH)3Ї
ZnCl2 + KOH = KCl + Zn(OH)Cl
Ca(HCO3)2 + Ca(OH)2 = 2 CaCO3 + 2 H2O.
9. Взаимодействие между двумя солями. Это один из самых распространённых методов получения солей. Из двух участвующих в реакции солей в результате двойного обмена образуются две новые соли. Реакции этого типа протекают до конца лишь в том случае, если один из продуктов удаляется из сферы реакции (выпадает в осадок):
BaCl2 + Na2SO4 = BaSO4Ї + 2 NaCl
Ag2SO4 + 2 KI = 2 AgIЇ + K2SO4.
10. Взаимодействие между металлом и солью. Реакции протекают при условии, что металл находится в ряду напряжений левее металла, входящего в состав исходной соли:
Fe + CuSO4 = FeSO4 + CuЇ
Cu + Hg(NO3)2 = Cu(NO3)2 + HgЇ
11. Взаимодействие металла с неметаллом. Этим методом получают соли бескислородных кислот:
2 Fe + 3 Cl2 = 2 FeCl3
Zn + S = ZnS.
12. Взаимодействие металла со щёлочью. Металлы, оксиды которых амфотерны, реагируют с водными растворами щелочей, выделяя водород и образуя соли:
Zn + 2 NaOH = Na2ZnO2 + H2
2 Al + 6 KOH = 2 K3AlO3 + 3 H2.
13. Взаимодействие неметалла со щёлочью. Галогены, сера и некоторые другие элементы взаимодействуют со щелочами, образуя две соли одновременно — бескислородную и кислородсодержащую:
Сl2 + 2 KOH = KCl + KClO + H2O
3 S + 6 NaOH = 2 Na2S + Na2SO3 + 3 H2O.
14. Взаимодействие неметалла с солью. Некоторые неметаллы способны соединяться с солями с образованием новых солей:
Сl2 + 2 KI = 2 KCl + I2
S + Na2SO3 = Na2S2O3.
15. Термическое разложение солей. При нагревании некоторых кислородсодержащих солей образуются новые соли, с меньшим содержанием кислорода или вообще его не содержащие:
2 KNO3 = 2 KNO2 + O2
2 KClO3 = 2 KCl + 3 O2
4 Na2SO3 = Na2S + 3 Na2SO4.
25
КРЕМНИЙ
Ближайший аналог углерода — кремний — является третьим (после кислорода и водорода) по распространенности элементом: на его долю приходится 16,7 % от общего числа атомов земной коры. Если углерод можно рассматривать как основной элемент для всей органической жизни, то кремний играет подобную же роль по отношению к твёрдой земной коре, так как главная часть её массы состоит из силикатных пород, обычно представляющих собой смеси различных соединений кремния с кислородом и рядом других элементов. Весьма часто встречается и свободная двуокись кремния (SiO2), главным образом в виде обычного песка.
Свободный кремний впервые получен в 1823 г. Природный элемент слагается из трёх изотопов — 28Si (92,2 %), 29Si (4,7) и 30Si (3,1). Его практический атомный вес даётся с точностью до ±0,001.
В основном состоянии атом кремния имеет строение внешней электронной оболочки 3s23p2 и двухвалентен. Возбуждение его до ближайшего четырёхвалентного состояния (3s3p3) требует затраты 397 кДж/моль, т. е. почти такой же энергии, как и в случае углерода. Его сродство к одному электрону оценивается в 142 кДж/моль. Небольшие количества кремния присутствуют практически во всех частях человеческого организма, причём наиболее богаты им лёгкие (0,65 мг на г сухой ткани). Относительно много кремния содержат волосы и ногти. Имеются указания на избыточное его накопление раковыми опухолями (с одновременным уменьшением его содержания в моче).
Природный SiO2 служит исходным сырьём для получения всех остальных соединений кремния. В элементарном состоянии он может быть получен восстановлением SiO2 при высокой температуре магнием. Реакция начинается при поджигании смеси тонко измельчённых веществ и протекает по схеме:
SiO2 + 2 Мg = 2 МgO + Si + 293 кДж.
Для освобождения от МgО и избытка SiО2 продукт реакции последовательно обрабатывают соляной и плавиковой кислотами.
Для получения больших количеств элементарного кремния обычно используется проводимая в электрической печи реакция по уравнению:
SiO2 + 2 C = 2 CO + Si
(что даёт продукт не выше 99%-ной чистоты). Такой кремний иногда применяется для выделения свободных металлов из их оксидов (силикотермия). Значительно более чистый Si получается при взаимодействии паров четырёххлористого кремния и цинка около 1000 °С по реакции:
SiCl4 + 2 Zn = 2 ZnCl2 + Si,
а ещё более чистый — термическим разложением SiH4 на элементы при температурах выше 780 °С.
Кремний часто получают в виде сплава с железом (ферросилиция) сильным накаливанием смеси SiO2, железной руды и угля. Сплавы, содержащие до 20% Si, могут быть, таким образом, изготовлены в доменных печах, более высокопроцентные — в электрических. Ферросилиций непосредственно используется для изготовления кислотоупорных изделий, так как уже при содержании 15% Si на металл не действуют все обычные кислоты, кроме соляной, а при 50% Si перестаёт действовать и HСI. Важнейшее применение ферросилиций находит в металлургии, где он употребляется для введения кремния в различные сорта специальных сталей и чугунов.
Свойства кремния сильно зависят от величины его частиц. Получаемый при восстановлении SiO2 магнием аморфный кремний представляет собой бурый порошок. Перекристаллизовывая его из некоторых расплавленных металлов (например, Zn), можно получить кремний в виде серых, твёрдых, но довольно хрупких кристаллов с плотностью 2,3 г/см3. Кремний плавится при 1410 и кипит при 2620 °С.
Кристаллический кремний является веществом, химически довольно инертным, тогда как аморфный значительно более реакционноспособен. С фтором он реагирует уже при обычных условиях, с кислородом, хлором, бромом и серой — около 500 °С. При очень высоких температурах кремний способен соединяться с азотом и углеродом. Он растворим во многих расплавленных металлах, причём с некоторыми из них (Zn, AI, Sn, Pb, Au, Ag и т. д.) химически не взаимодействует, а с другими (Мg, Ca, Cu, Fe, Pt, Bi и т. д.) образует соединения (например, Мg2Si), называемые силицидами.
В кристаллическом состоянии кремний хорошо проводит тепло. Его электропроводность составляет 0,007 (для обычного) — 1·10-6 (для особо чистого) от электропроводности ртути, причём при нагревании она не понижается, а повышается. Повышается она и с увеличением давления, а при 120 тыс. атм кремний приобретает свойства металла. Теплота плавления кремния 46, теплота атомизации — 451 кДж/моль. Плавление сопровождается увеличением плотности (приблизительно на 9%), т.е. кремний в этом отношении подобен льду. Резко (в 20 раз) возрастает при плавлении и электропроводность кремния.
Кремний кристаллизуется по типу алмаза [d(SiSi) = 235 пм]. Его монокристаллы получают выращиванием в вакууме из расплава (путём медленного вытягивания соприкасающейся с поверхностью жидкости затравки). Таким путём удавалось выращивать монокристаллы диаметром 2,5 см и длиной 24 см. Подобные монокристаллы из очень чистого кремния с соответственно подобранными добавками служат для изготовления различных полупроводниковых устройств (выпрямителей переменного тока и др.).
Важное место среди таких устройств занимают фотоэлементы, служащие для прямого преобразования световой энергии в электрическую. Максимум их поглощения приходится на инфракрасные лучи. Коэффициент полезного действия кремневых фотоэлементов составляет около 15%. Из них построены, в частности, солнечные батареи, обеспечивающие питание радиоаппаратуры на искусственных спутниках Земли. В будущем рисуется перспектива массового наземного применения таких батарей для эффективного использования солнечной энергии (которой Земля ежегодно получает примерно в 100 раз больше, чем могло бы дать сжигание всех известных запасов ископаемого топлива).
При нагревании газообразный фтористый водород реагирует с кремнием по схеме:
Si + 4 HF = SiF4 + 2 H2.
Выше 300 °С на мелко раздробленный кремний начинает действовать НСl, а выше 500 °С — НBr. В обоих случаях образуется смесь водорода с SiГ4 и водородгалогенидными производными кремния (SiHГ3, SiH2Г2, SiH3Г).
Подобно карбидам, для силицидов известны только простейшие формулы. Иногда они согласуются с обычными валентностями образующих их металлов и кремния (например, Мg2Si, Mn2Si, MnSi, ReSi), но в большинстве случаев валентные соотношения остаются неясными (например, Mn3Si, Mn5Si3, MnSi2, Re3Si, ReSi2, Cr3Si, Cr5Si3, CrSi, CrSi2, Mo3Si, MoSi2, W5Si3, W3Si2, WSi2, V3Si, VSi2, Nb2Si, Nb5Si3, NbSi2, Ta2Si, Ta5Si3, TaSi2).
Как правило, силициды характеризуются большой твёрдостью и устойчивостью по отношению к нагреванию (например, Мg2Si плавится при 1085, МnSi — при 1270, а ТаSi2 — при 2200 °С). Многие из них очень устойчивы и по отношению к окислению при высоких температурах (например, ReSi2 — до 1600, а МоSi2 — до 1800 °С). Дисилицид молибдена используется в качестве защитного покрытия изделий из молибдена. Некоторые силициды (например, ReSi2, CrSi2) могут быть использованы как высокотемпературные полупроводники.
Лишь силициды более активных металлов (в частности, Li3Si, CaSi, CaSi2, Mg2Si) разлагаются водой и разбавленными кислотами, а большинство остальных по отношению к этим реагентам очень устойчиво. Напротив, щелочами многие силициды (особенно с большим содержанием Si) довольно легко разлагаются.
Кислоты на кремний при обычных условиях не действуют (за исключением смеси НF + HNO3). Щёлочи с выделением водорода переводят его в соли кремневой кислоты (Н2SiO3):
Si + 2 NaOH + H2O = Na2SiO3 + 2 H2.
Такая реакция протекает даже со слабыми щелочами.
Для возможности взаимодействия кремния со щелочами достаточно уже настолько ничтожных концентраций ионов ОН-, что реакция медленно идёт даже с водой, содержащей только следы щелочей, извлечённых из стекла. Так как образующаяся соль очень слабой кремневой кислоты в растворе практически нацело гидролизована, концентрация ионов ОН- по мере протекания реакции не уменьшается. Поэтому рассматриваемый процесс практически сводится к разложению воды кремнием, причём присутствующая в виде следов щёлочь играет роль катализатора. Для получения подобным образом 1 м3 водорода требуется затратить только 0,63 кг кремния, тогда как, напротив, железа потребовалось бы 2,5 кг (да к тому же ещё большее количество необходимой для реакции кислоты).
Очень химически активный элементарный кремний может быть получен действием при 30 °С хлора (без избытка) на взвесь СаSi2 в ССI4 по реакции:
СаSi2 + Cl2 = CaCl2 + 2 Si.
Такой кремний бурно реагирует не только с водой, но и с СН3ОН.
Подобно свободному кремнию реагируют со щелочами и многие силициды, в частности силицид железа. Особенно удобна для быстрого получения водорода в полевых условиях смесь порошка высокопроцентного ферросилиция с сухими Са(ОН)2 и NaOH. При поджигании она начитает тлеть с энергичным выделением водорода по схеме:
Si + Ca(OH)2 + 2 NaOH = Na2SiO3 + CaO + 2 H2.
Смесь эта носит техническое название гидрогенит.
Наиболее характерным и устойчивым соединением кремния является его диоксид (SiO2), образование которого из элементов идёт с очень большим выделением тепла:
Si + O2 = SiO2 + 911 кДж.
Диоксид кремния представляет собой бесцветное, очень тугоплавкое твёрдое вещество.
Каждый атом кремния в кристаллах SiO2 тетраэдрически окружён четырьмя атомами кислорода [d(SiO) = 161 пм], а каждый атом кислорода является одновременно составной частью двух тетраэдров. Плоская схема такой полимерной структуры показана на рисунке (1.1).
Основной природной формой диоксида кремния является минерал кварц (плотность 2,65 г/см3, показатель преломления 1,55). Гораздо реже встречаются характеризующиеся несколько иными кристаллическими структурами и меньшей плотностью минералы тридимит и кристобалит. При медленном нагревании кварца сначала (573 °С) происходит некоторое изменение его собственной кристаллической структуры (a-кварц ® b-кварц), после чего он последовательно переходит в две другие формы и лишь затем плавится:
867 1470 1723
кварц ® тридимит ® кристобалит ® жидкий SiO2.
Быстрым нагреванием можно расплавить кварц при 1610 °С, а тридимит — при 1680 °С. У кристобалита существует и низкотемпературная модификация (ниже 272 °С), а у тридимита — даже восемь таких модификаций (все ниже 475 °С). Для точки кипения диоксида кремния даётся значение 2590 °С (но в высоком вакууме кремнезём заметно испаряется уже выше 1200 °С). Пары диоксида кремния сильно диссоциированы по схеме:
SiO2Ы SiO + O
Энергия такой диссоциации оценивается в 468 кДж/моль.
| | Под высоким давлением могут быть получены ещё
ѕ Si—O—Si— две кристаллические модификации SiO2 — коэсит
| | (плотность 2,9 г/см3, показатель преломления 1,60)
O O и стишовит (плотность 4,3 г/см3, показатель
| | преломления 1,80). По внутренней структуре
ѕ SiѕOѕSiѕ кристалла коэсит отличается от кварца лишь иным
| | взаимным расположением сопряжённых друг с
Рис 1.1. Схема структуры другом тетраэдров SiO4, т.е. характерная для кремния
диоксида кремния. тетраэдрическая координация атомами кислорода в
нём сохраняется. Напротив, в стишовите имеет место совершенно необычная для кислородных соединений кремния октаэдрическая координация его атома. По твёрдости рассматриваемые формы SiO2 располагаются в ряд: стишовит > коэсит > кварц. Область устойчивости коэсита лежит выше 20 атм, стишовита — выше 100 атм. Под обычным давлением стишовит переходит в кварц при 400 °С, а коэсит — при 1200 °С.
Кристаллы кварца иногда встречаются громадной величины (самый крупный, массой около 70 т, был обнаружен в 1958 г. в Казахстане). Они вращают плоскость поляризации света, причём могут быть право- и левовращающими.
Свободный диоксид кремния (иначе кремнезём, кремневый ангидрид) встречается главным образом в виде минерала кварца. Загрязнённый примесями кварц — обычный песок — является одним из основных продуктов разрушения горных пород и одновременно одним из важнейших строительных материалов, ежегодное мировое потребление которого исчисляется сотнями миллионов тонн. На долю свободного диоксида кремния приходится приблизительно 12% от массы всей земной коры. Гораздо большее количество SiO2 (около 43% от массы земной коры) химически связано в составе различных горных пород. В общем, следовательно, земная кора более чем наполовину состоит из диоксида кремния.
Некоторые минералогические разновидности кварца носят особые названия. Так, его большие прозрачные кристаллы часто называют горным хрусталем, окрашенную в фиолетовый цвет разновидность —аметистом. К мелкокристаллическим модификациям кремнезёма (с примесями других веществ) относятся агат и яшма.
Кварц используется в различных областях техники, и большие его кристаллы часто выращивают искусственно. Это обычный исходный материал для конструирования аппаратуры для получения ультразвуковых волн. Применимость кварца в этой области основана на его пьезоэлектрических свойствах — особом отношении вырезанной из кристалла пластинки к быстропеременному электрическому полю: под его действием пластинка начинает периодически сжиматься и расширяться с частотой, равной частоте наложенного поля. Благодаря этому в окружающей пластинку среде возбуждаются волны, аналогичные обычным звуковым, но характеризующиеся иной частотой.
Человеческое ухо воспринимает звуковые волны с частотами в пределах примерно от 16 герц до 20 тыс. герц (колебаний в секунду). Звуки с частотами более низкими (инфразвуки) и более высокими (ультразвуки) нашему непосредственному восприятию недоступны. Наиболее слабые звуки воспринимаются нами в области около 3 тыс. Гц. Звуки выше известной силы не воспринимаются как таковые, а вызывают болевые ощущения. Обычный звуковой интервал человеческой речи составляет от 120 Гц до 400 Гц, а используемый в музыке — от 50 Гц до 8 тыс. Гц. Самый низкий певческий голос имеет частоту 80 Гц, а самый высокий — 1300 Гц.
Сила звука обычно оценивается по шкале децибелов (дб), в которой нулевая отметка соответствует самому слабому звуку, воспринимаемому нормальным ухом. Представление об этой (имеющей логарифмический характер) шкале дают следующие её средние оценки (в дб): нормальное дыхание (10), шёпот (25), разговорная речь (60), среднее уличное движение (70), поезд метро (95), реактивный самолёт на высоте 150 м (115), порог болевой чувствительности человека (125). Шум городского дома оценивается в 30 ч 55 дб. Считается, что постоянный шум с уровнем более 85 дб может привести к частичной потере слуха.
Некоторые животные только потому и кажутся нам “немыми”, что используемый ими интервал частот лежит вне пределов слышимости человека. Установлено, например, что рыбы оживлённо переговариваются друг с другом, причём отдельным видам соответствуют различные говоры. Благодаря тому, что вода мало поглощает звук, а скорость его распространения в ней велика (около 1500 м/с), эти “рыбьи разговоры” могут происходить на больших расстояниях. У дельфинов максимум интенсивности испускаемых звуков приходится на интервал 20 ч 60 кГц, но диапазон их возможного восприятия находится гораздо шире (18 Гц ч 280 кГц). Известно также, что ориентировка летучих мышей при полёте основана на испускании ими ультразвуков и восприятии их отражений от окружающих предметов. За секунду испускается до 60 звуковых импульсов с наиболее интенсивными частотами в пределах 35—70 тыс. Гц. Улавливание этих звуковых импульсов ночными бабочками помогает им спасаться от летучих мышей. Может быть сконструирован свисток, сигналы которого слышит собака (воспринимающая звуки до 100 тыс. Гц), но не слышит человек.
В настоящее время удаётся возбуждать ультразвуковые волны с частотами порядка десятков миллиардов герц. Подобно обычному звуку, ультразвуковые волны можно собирать и направлять на определённые объекты при помощи рефлекторов. Энергия звуковых колебаний растёт пропорционально квадрату их частоты. Имеется установка, способная создавать интенсивность ультразвука более 100 кВт/см3.
Короткие ультразвуковые волны обладают рядом интересных свойств. Они разрушают многие сложные молекулы, убивают мелких рыб, стимулируют прорастание семян, позволяют получать устойчивые эмульсии, вызывают протекание некоторых химических реакций. Основной причиной всех этих эффектов являются резкие местные колебания давления и температуры, обусловленные быстропеременным возникновением и исчезновением пустот (“кавитаций”) в подвергаемой действию ультразвука среде.
При помощи ультразвуковых волн можно легко и удобно контролировать однородность толстых механических блоков, производить разнообразную механическую обработку самых твёрдых материалов (вплоть до алмаза), пайку трудно спаиваемых металлов (например, алюминия), мойку шерсти, создавать эхолоты для измерения морских глубин, гидролокаторы для обнаружения косяков рыб и т. д. Был сконструирован ультразвуковой микроскоп, позволяющий получить изображение предметов, находящихся в непрозрачных средах, с увеличением до нескольких тысяч раз. Частота 19,5 кГц оказалась непереносимой для крыс, и генератор мощностью всего в 35 Вт надёжно освобождает от них площадь 225 м2.
Хуже изучены инфразвуки, которые присутствуют во всех шумах (атмосферы, моря, леса, городского движения, работающих моторов и др.). Инфразвуки хорошо распространяются в воздухе на громадные расстояния. Так, улавливая возникающие при трении волн о воздух инфразвуки с частотами 8—13 Гц, морские животные заранее узнают о приближении шторма. Уже создан электронный прибор, работающий на том же принципе. Делаются также успешные попытки использовать инфразвуки для медицинского “прозвучивания” человеческого тела. Вместе с тем выяснилось, что инфразвуки повышенной мощности (особенно в области 6 ч 9 Гц) оказывают вредное влияние на организм. Обусловлено это их резонансным наложением на собственные колебания внутренних органов человека. Особенно опасна частота 7 Гц, так как она совпадает с частотой a-ритма биотоков мозга.
Резонансное наложение инфразвуков на собственные колебания материальных объектов может привести к их разрушению. Так, сообщалось, что при включении генератора звука с частотой 3,5 Гц и мощностью 100 Вт стены лаборатории угрожающе затряслись, потолок покрылся трещинами, и опыт пришлось прекратить.
На основе SiO2 готовится важный огнеупорный материал — динас. Его получают обжигом при 1300—1400 °С измельчённого кварца, к которому добавлено 2—2,5% извести. Динасовый кирпич размягчается лишь около 1700 °С и служит, в частности, для выкладки сводов мартеновских печей.
Известен также оксид кремния(II). В природе он не встречается, но может быть получен по реакции:
SiO2 + Si = 2 SiO.
Под обычным давлением возгонка монооксида кремния начинается около 1200 °С (когда сами исходные вещества ещё практически не испаряются). В парах SiO является индивидуальным соединением. Энергия диссоциации на элементы 789 кДж/моль. Перевод его в твёрдое состояние может быть осуществлён только быстрым охлаждением (“закалкой”) газовой фазы. В противном случае успевает пройти дисмутация по уравнению:
2 SiO = SiO2 + Si.
Получающаяся твёрдая фаза представляет собой чрезвычайно мелкий коричневый порошок. Монооксид кремния медленно окисляется кислородом воздуха и легко растворяется в щелочах с образованием солей кремневой кислоты и выделением водорода. Он легко электризуется от трения, приобретая сильный отрицательный заряд.
Нитрид кремния (Si3N4). Прямой синтез происходит лишь выше 1300 °С, но сопровождается значительным выделением тепла (748 кДж/моль). Нитрид кремния представляет собой лёгкий белый порошок, около 1900 °С возгоняющийся. Он известен в двух кристаллических формах [d(SiN) = 172—175 пм] и очень устойчив по отношению к различным химическим воздействиям. Так, расплавленные щёлочи медленно растворяют его по схеме:
Si3N4 + 12 NaOH = 3 Na4SiO4 + 4 NH3,
но раствор NaOH не действует даже при кипячении. До 1000 °С нитрид кремния не реагирует ни с О2, ни с Н2, ни с водяным паром. Горячая концентрированная плавиковая кислота разлагает его по схеме:
Si3N4 + 16 HF = 2 (NH4)2SiF6 + SiF4
лишь крайне медленно, а концентрированная НСI вообще не действует. Под высоким давлением в атмосфере азота нитрид кремния хорошо прессуется и спекается (при 1500 °С). Около 1000 °С он приобретает полупроводниковые свойства.
Сообщалось о получении (довольно сложным косвенным путём) и другого нитрида кремния — Si2N2, который представляет собой белый рентгеноаморфный порошок, при нагревании выше 1200 °С переходящий в Si3N4. Взаимодействием SiCl4 с жидким аммиаком был получен полимерный имид кремния — [Si(NH)2]n.
Нагреванием Si и SiO2 в атмосфере азота при 1450 °С был получен оксонитрид Si2N2O, простейшей формуле которого отвечает строение O(SiN)2.
Жёлто-коричневый фосфид кремния может быть получен взаимодействием элементов (выше 700 °С). Это игольчатые кристаллы с красным металлическим блеском, отвечающие формуле SiP. Синтезом из элементов был получен и чёрный SiP2.
При прокаливании смеси SiO2 c углём в электрической печи до 2000 °С образуетсякарбид кремния (SiC), называемый обычно карборундом. Реакция идёт по суммарному уравнению:
SiO2 + 3 C + 527 кДж = 2 СО + SiC
и требует затраты около 8 тыс. кВт·ч на тонну SiC. Чистый карборунд представляет собой бесцветные кристаллы (выше 2200 °С разлагающиеся на элементы), а технический продукт обычно окрашен примесями в тёмный цвет. Ежегодная мировая выработка карборунда составляет около 100 тыс. т.
Из свойств карборунда наиболее важна его твёрдость, уступающая лишь твёрдости алмаза. Он широко применяется для выработки твёрдых материалов. В частности, из него обычно изготовляют круги точильных станков. Карборунд обладает хорошей теплопроводностью и полупроводниковыми свойствами (n-типа), которые сохраняются до 1000 °С (тогда как у элементарного кремния они теряются уже выше 250 °С). Он находит использование для изготовления электропечей, однако для этих целей чаще применяют силит, получаемый обжигом при 1500 °С (в атмосфере СО или N2) массы, сформированной из смеси карборунда, кремния и глицерина. Силит обладает механической прочностью, химической стойкостью и хорошей электропроводностью.
С химической стороны карборунд характеризуется своей индифферентностью по отношению ко всем обычным кислотам (кроме смеси концентрированных HF и HNO3). Напротив, при сплавлении со щелочами в присутствии воздуха он легко разлагается с образованием солей кремневой и угольной кислот. Выше 800 °С карборунд начинает заметно окисляться кислородом воздуха, а выше 1300 °С реагирует с водяным паром по схеме:
SiC + 2H2O = SiO2 + CH4.
Хлорирование SiC (при температурах выше 600 °С) ведёт к образованию SiCI4 и свободного углерода.
В воде SiO2 практически нерастворим. Не действуют на него и кислоты, за исключением HF, которая реагирует по схеме:
SiO2 + 4 HF = SiF4 + 2H2O.
Щёлочи постепенно переводят SiO2 в раствор, образуя соответствующие соли кремневой кислоты (силикаты), например, по реакции:
SiO2 + 2 NaOH = Na2SiO3 + H2O.
Очень мелко раздробленный диоксид кремния быстро растворяется при кипячении с растворами щелочей, обычно же реакцию получения кремнекислых солей проводят путём сплавления SiO2 со щелочами или соответствующими карбонатами, из которых при высокой температуре выделяется СО2, например, по схеме:
SiO2 + Na2CO3 = Na2SiO3 + CO2.
В результате реакция сводится к выделению угольной кислоты кремневой кислотой.
Кристаллы Na2SiO3 (т. пл. 1027 °С) слагаются из цепей типа [–OSi(O2)–]n, в промежутках между которыми располагаются ионы натрия.
Гидролиз метасиликата натрия идёт с образованием двуметасиликата по схеме:
2 Na2SiO3 + H2O = Na2Si2O5 + 2 NaOH,
причём в нормальном растворе гидролизовано 14, в 0,1 н. — 28 и в 0,01 н.— 32%. Гидролиз двуметасиликата идёт уже значительно слабее. Так, в нормальном растворе гидролизуется его 2,4, в 0,1 н. — 6%.
Наряду с Na2SiO3 в растворимом стекле содержатся и более сложные силикаты натрия. Их обычный состав может быть выражен формулой Na2O·nSiO2, где n = 2 ё 4. Постепенным отщеплением части SiO2 обусловлены те изменения в жидком стекле (помутнение, а иногда и застывание нацело в студнеобразную массу), которые часто наблюдаются при долгом его хранении. Жидкое стекло следует держать в сосудах с резиновыми пробками (так как стеклянные и корковые сильно приклеиваются к горлышку).
Производство жидкого стекла достигает значительных размеров (порядка сотен тысяч тонн ежегодно), так как оно используется для укрепления грунтов при строительных работах и в ряде различных отраслей промышленности. Пропитка им бетонных автомобильных дорог значительно увеличивает их сопротивление истиранию. Ввиду того, что пропитанные жидким стеклом изделия из дерева и тканей очень трудно загораются, подобной пропитке часто подвергают, например, материалы, идущие для изготовления театральных декораций. Силикат натрия входит в состав некоторых стиральных порошков. Опущенные в его разбавленный раствор свежие яйца могут длительное время сохраняться при обычной температуре.
Жидкое стекло непосредственно используется в качестве конторского клея и часто служит основой огнеупорных замазок. Простая по составу замазка, пригодная для склеивания стекла и фарфора, может быть получена замешиванием отмученного мела с жидким стеклом до консистенции теста. Последнее довольно быстро затвердевает в очень прочную массу белого цвета. Быстро твердеющая замазка из замешанного на жидком стекле цемента пригодна для склеивания камней.
Как правило, силикаты (кремнекислые соли) бесцветны, тугоплавки и практически нерастворимы в воде. К числу немногих растворимых относится Na2SiO3 — “растворимое стекло”.
Кремневая кислота очень слабая, поэтому “жидкое стекло” показывает в результате гидролиза резко щелочную реакцию, а силикаты слабых оснований гидролизованы в растворе практически нацело. По той же причине кремневая кислота выделяется из растворов своих солей многими другими кислотами, в том числе угольной.
Если в растворе угольная кислота выделяет кремневую из её солей, то при прокаливании идёт обратная реакция. Первая обусловлена меньшей силой (степенью диссоциации) кремневой кислоты, вторая — её меньшей летучестью при нагревании. Ряд кислот по их сравнительной летучести может отличаться от ряда тех же кислот по их силе, направления реакций выделения в растворе с одной стороны и при прокаливании — с другой могут быть различными, что видно из схемы:
выделение в раствореЮ
HCl H2SO4 H3PO4 H2SiO3
Ьвыделение при прокаливании.
Свободная кремневая кислота почти нерастворима в воде (в форме истинного раствора). Однако она легко образует коллоидные растворы и поэтому обычно осаждается только частично. Осадок имеет вид бесцветного студня, причём состав его отвечает не простой формуле Н2SiO3 или Н4SiO4, а более общей — nSiO2·m H2O cо значениями n и m, изменяющимися в зависимости от условий осаждения. Значениям n>1 соответствуют различные поликремневые кислоты, производными которых по химическому составу могут считаться многие минералы.
Истинная растворимость кварца в воде очень мала (причём растворение протекает крайне медленно), но аморфного кремнезёма — значительно выше. Вследствие взаимодействия по схемам:
SiO2 + OH’ Ы HSiO3’и НSiO3’ + H2O Ы H2SiO3 + OH’
даже следы щелочей сильно способствуют переходу SiO2 в жидкую фазу (с образованием коллоидного раствора). Поэтому хранящиеся в стеклянных сосудах водные растворы часто содержат следы кремнезёма.
Из отдельных гидратных форм кремневой кислоты (отвечающих различным значениям n и m общей формулы) возможность существования в качестве определённых соединений более или менее надёжно установлена для метакремневой кислоты — Н2SiO3 (n = 1, m = 1), двуметакремневой — Н2Si2O5 (n = 2, m = 1), ортокремневой — Н4SiO4 (n = 1, m = 2) и кислоты Н6Si2O7 (n = 2, m = 3). Основной формой существования свободной кремневой кислоты в растворе является Н4SiO4 (К1 = 3·10-10, К2 = 2·10-12). В результате её полимеризации с последующим отщеплением молекул воды могут образовываться разнообразные поликремневые кислоты.
Интересные результаты были получены при изучении взаимодействия SiO2 с водяным паром при высоких температурах (400-700 °С) и давлениях (5-500 атм). В зависимости от плотности пара существуют три основных типа такого взаимодействия. До 0,05 г/см3 реакция идёт с образованием в газовой фазе Si(OH)4 (т.е. SiO2·2H2O), в интервале 0,1-0,45 образуется (ОН)3SiOSi(OH)3 (т.е. 2SiO2·3H2O), а выше 0,65 г/см3 — Н2SiO3 (т.е. SiO2·H2O).
Природные формы кремнезёма с содержанием n » m встречаются в виде неорганических образований — кремня, опала, трепела и др., а также остатков панцирей некогда живших мельчайших морских организмов — диатомита (“инфузорной земли”). На чрезвычайной пористости трепелов и диатомитов основано их применение для изготовления динамита, для очистки масел и в качестве изоляционного материала. Кремень является одним из важнейших в истории человечества минералов, так как он служил основным материалом для производства орудий труда в каменном веке и затем на протяжении многих столетий с ним было неразрывно связано получение огня. Получаемая из кремневой кислоты аморфная двуокись кремния (“белая сажа”) находит использование в резиновой промышленности, а также при производстве смазок, красок и лаков.
Образование пероксидных соединений для кремния не характерно, и производные надкислот этого элемента не получены. Вместе с тем известны продукты присоединения перекиси водорода к SiO2 и солям кремневой кислоты, например Na2H2SiO4·2H2O2. Вещество это представляет собой белый порошок, легкорастворимый в воде.
В противоположность широко распространённым кислородным производным кремния, его сернистые соединения в природе не встречаются. Теплота образования SiS2 из элементов равна 208 кДж/моль. Кремнийдисульфид может быть получен при 1300 °С по реакции:
2 H2S + Si = SiS2 + 2 H2
и выделяется в виде белых шелковистых игл (т. пл. 1090, т. кип. 1130 °С). Иглы эти слагаются из полимерных цепей. Водой SiS2 разлагается на SiO2 и H2S, а с растворимыми сульфидами образует соли тиокремневой кислоты (H2SiS3). Как и в случае углерода, эфиры тиокремневой кислоты известны и для её орто-формы [например, Si(SC2H5)4]. Взаимодействием SiS2 с избытком Si (в вакууме при 850 °С) может быть получен односернистый кремний (SiS)n, представляющий собой жёлтые иглы, разлагающиеся водой с выделением Н2S и Н2.
Соли кремневых кислот известны для гидратных форм с самыми различными значениями n и m. Продуктами полного или частичного замещения в них водорода на те или иные металлы являются так называемые простые силикаты. Примером их может быть минерал асбест (Mg3H4Si2O9 или 3MgO·2H2O·2SiO2).
Значительно более распространены в природе сложные силикаты, производящиеся главным образом от кислот общей формулы хЭ2О3·уSiO2·zH2O. Важнейшими соединениями этого типа являются алюмосиликаты (где Э — АI), особенно относящихся к группе полевых шпатов, на долю которых приходится более половины массы земной коры. Минералы:
ортоклаз К2Аl2Si6O16 или К2О·Аl2O3·6SiO2
альбит Na2Al2Si6O16 или Na2O·Al2O3·6SiO2
анорит СаАl2Si2O8 или СаО·Аl2O3·2SiO2
могут быть названы в качестве основных их представителей.
В зависимости от типа исходного гидрата кремнезёма простые силикаты иногда подразделяются на метасиликаты (производные H2SiO3), ортосиликаты (производные H4SiO4) и силикаты с иным соотношением n и m. Важным простым силикатом является тальк (Mg3H2Si4O12 или 3MgO·H2O·4SiO2). Обладающий очень высокой механической прочностью зелёный минерал нефрит (Ca2Э5H2Si8O24 или 2CaO·5ЭO·H2O·8SiO2, где Э — Mg и Fe в переменных количествах) нередко применялся в каменном веке для изготовления орудий труда.
К весьма распространённым алюмосиликатам относятся, в частности, минералы группы слюд, например мусковит (калийная слюда) — К2H4Al6Si6O24 или K2O·2H2O·3Al2O3·6SiO2. Большое техническое значение имеет алюмосиликат нефелин (Na2Al2Si2O8 или Na2O·Al2O3·2SiO2), используемый для комплексной выработки из него глинозёма (Al2О3), соды и цемента.
Драгоценным камнем является топаз [Al(F,OH)]2SiO4, кристаллы которого могут обладать разнообразной окраской светлых оттенков и достигать иногда гигантских размеров (масса наибольшего равнялась 117 кг).
Горные породы очень часто представляют собой смеси различных минералов, что заметно уже по их внешнему виду. Например, гранит состоит из смеси кристаллов кварца, полевых шпатов и слюд, причём общее содержание SiO2 в нём составляет около 70%.
Интересную особую группу алюмосиликатов образуют цеолиты, состав которых может быть выражен формулой МxЭyO2y·nH2O, где М — Ca, Na (реже Ba, Sr, K), а Э — Si, Al в переменных соотношениях. Они обладают рыхлой кристаллической структурой, образованной имеющими общие атомы кислорода тетраэдрами SiO4 и AlO4, в пустотах которой располагаются катионы М и молекулы воды. Цеолиты способны обменивать содержащуюся в них воду на другие жидкости (спирт, аммиак и т. п.), а катионы М — на различные другие катионы. В отличие от конституционной (т. е. входящей в основной состав вещества) воды асбеста, талька, мусковита и ряда других минералов, т. н. цеолитная вода ведёт себя, как сорбированная. При осторожном нагревании цеолитов она удаляется постепенно, причём даже полное обезвоживание не ведёт к разрушению основной структуры минерала.
Важной разновидностью цеолитов являются т. н. молекулярные сита, характеризующиеся наличием узких сквозных пустот в их структуре. Природные цеолиты этого типа встречаются редко, и обычно их вырабатывают искусственно (из Na2SiO3, NaAlO2 и NaOH). Получаемые кристаллы имеют однородные по размерам отверстия диаметром большей частью от 300 до 1300 пм и активно сорбируют молекулы (особенно полярные), которые могут в эти отверстия войти. Например, молекулярное сито с диаметром отверстий 350 пм поглощает Н2, О2 и N2, но практически не поглощает Ar или СН4. Подобное же различие при больших размерах отверстий проявляется и по отношению к сложным молекулам. Тем самым создаётся возможность избирательного извлечения некоторых веществ из смесей, их освобождения от вредных примесей и т. п. В частности, молекулярными ситами пользуются для осушки некоторых газов и жидкостей. Они применяются также в качестве ионообменников и носителей катализаторов.
Пространственное строение многих силикатов было изучено с помощью рентгеновских лучей. При этом выяснилось, что все исследованные структуры могут быть классифицированы с разбивкой на сравнительно небольшое число типов, отличающихся друг от друга характером сочетания тетраэдрических ионов SiO44-.
Некоторые из таких типов включают в себя простейшие силикатные анионы. Сюда относятся прежде всего случаи заполнения узлов пространственной решётки индивидуальными ионами SiO44-. Второй тип характеризуется наличием в узлах решётки ионов Si2O76-(образованных двумя тетраэдрами SiO44- c одним общим углом), остальные — наличием в узлах решётки циклических ионов Si3O96-, Si4O128- или Si6O1812- (образованных тетраэдрами SiO44- c двумя общими углами у каждого из них).
Другие типы силикатных структур могут быть названы групповыми, так как они слагаются из теоретически бесконечного числа тетраэдров SiO44-. Такие сочетания могут иметь характер простой цепи, двойной цепи или плоскости. Наконец, существуют типы, представляющие собой объёмную структуру. Во всех подобных решётках часть ионов Si4+ может быть заменена на ионы Al3+ и др., а часть ионов O2- на ионы ОН- и др. Вместе с тем часть входящих в состав силиката ионов (K+, Na+ и др.) может располагаться между цепями или плоскостями, а также в промежутках трёхмерной структуры.
Нагреванием силицидов Са, Mg и Li в парах серы были синтезированы некоторые простые тиосиликаты этих металлов — CaSiS3, CaSi2S5, Ca2SiS4, Mg2SiS4, Li2SiS3, Li4SiS4 (синтезирован также Ca2SiSe4). Oни представляют собой бесцветные кристаллические вещества, легко подвергающиеся гидролизу.
Под совместным действием различных природных факторов, главным образом воды и углекислого газа, природные силикаты, алюмосиликаты и т. п. на земной поверхности постепенно разрушаются (“выветриваются”), причём растворимые продукты уносятся водой в океан, а нерастворимые частично отлагаются на месте, частично оседают в руслах рек или выносятся в море. Основными нерастворимыми продуктами распада наиболее распространённых в природе алюмосиликатов являются кварц (SiO2), оседающий в виде песка, и каолин (Н4AI2Si2O9 или AI2O3·2SiO2·2H2O), представляющий собой основу обычных глин (окрашенных в бурый цвет примесями железа), а в более чистом состоянии образующий иногда залежи белой глины. Процесс выветривания алюмосиликата может быть изображён следующей схемой:
К2АI2Si6O16 + 2 H2O + CO2 = K2CO3 + H4AI2Si2O9 + 4 SiO2 ортоклаз каолин кварц
Песок и глина создают минеральную основу всех видов почв. Характер последних зависит в основном от условий температуры и влажности данной местности.
В действительности процесс проходит, по-видимому, через следующие стадии:
K2Al2Si6O16 + H2CO3 = K2CO3 + H2Al2Si6O16 и
H2Al2Si6O16 + 14 H2O = 2 Al(OH)3 + 6 H4SiO4, затем
2 Аl(OH)3 + 2 H4SiO4 = 5 H2O + H4Al2Si2O9 и
4 H4SiO4 = 8 H2O + SiO2.
Уравнения показывают, что под действием угольной (или какой-либо другой) кислоты и воды первоначально происходит полное разрушение алюмосиликата, после чего из остатков формируется каолин и постепенно выкристаллизовывается кварц.
Из получаемых искусственно нерастворимых в воде силикатов наиболее важным является стекло, известное человечеству ещё с глубокой древности.
Состав “нормального” стекла выражается формулой Na2CaSi6O14 или Na2O·CaO·6SiO2. Довольно близко к этому составу подходит обычное оконное стекло. Путём частичной замены Na, Ca и Si на другие элементы удаётся получать различные специальные сорта, характеризующиеся теми или иными требуемыми для отдельных применений качествами.
Основными продуктами стекольного производства являются сода, известняк и песок. Процесс образования “нормального” стекла может быть выражен уравнением:
Na2CO3 + CaCO3 + 6 SiO2 = 2 CO2 + Na2O·CaO·6SiO2.
Смесь исходных веществ нагревают приблизительно до 1500 °С и выдерживают расплавленную массу до полного удаления газов, после чего она пускается в дальнейшую переработку.
Хотя стекло в целом практически нерастворимо, однако вода частично разлагает его с поверхности, вымывая преимущественно натрий. Подобно воде действуют и кислоты (кроме плавиковой) — стекло, находящееся некоторое время в соприкосновении с водой или кислотами, дальше практически не разрушается ими. Напротив, из-за сильного преобладания SiO2 в составе стекла действие на него щелочей имеет длительный характер. Поэтому хранящиеся в стеклянных сосудах жидкости обычно содержат примеси растворимых силикатов.
При выработке стекла соду нередко заменяют более дешевой смесью сульфата натрия и угля. В этом случае идёт реакция:
Na2SO4 + C + CaCO3 + 6 SiO2 = Na2O·CaO·6SiO2 + SO2 + CO2.
Реакция взаимодействия “нормального” стекла с HF может быть выражена схемой:
Na2CaSi6O14 + 28 HF = 2 NaF + CaF2 + 6 SiF4 + 14 H2O.
Аналогично идёт реакция и при действии HF на другие стёкла, а также природные силикаты. Из последних наиболее близки по составу к стеклам некоторые вулканические лавы.
Стёкла иенское и пирекс характеризуются большой устойчивостью по отношению к действию воды и кислот, а также сравнительно малыми (особенно пирекс) коэффициентами расширения, вследствие чего они хорошо переносят нагревание. Из них изготовляют высококачественную химическую посуду для лабораторий. Так как стекло типа пирекс отличается также большой механической прочностью, из него в настоящее время делают не только предметы домашнего обихода, но и сосуды для промышленного проведения химических реакций. Ввиду малого коэффициента расширения сосуды эти можно нагревать непосредственно на открытом огне.
Анализ бесцветного и прозрачного древнеегипетского стекла (около 1500 лет до н. э.) дал следующие результаты (в вес. %): SiO2—63,4; Na2O — 22,4; K2O — 0,8; CaO — 7,8; MgO — 4,2; Al2O3 — 0,6; Fe2O3 — 0,7; MnO — следы. Стекло было известно в Египте ещё за 1500 лет до н. э.
Примеси различного типа придают стеклу различные цвета. Так соли двухвалентного железа сообщают стеклу зеленую окраску (“бутылочное стекло”). Для её уничтожения обычно добавляют Se, растворы которого в стекле имеют розовый цвет.
Иногда примеси искусственно вводят для получения цветного стекла. Так, соединения кобальта окрашивают стекло в синий цвет, Cr2O3 — в изумрудно-зелёный, соединения марганца — в фиолетовый и т. д. В других случаях добиваются изменения каких-либо специальных свойств стекла. Например, стекло, содержащее в своём составе СdO, задерживает нейтроны, PbO — рентгеновские лучи, а оксиды ванадия — ультрафиолетовые лучи.
Последние в большей части задерживаются и обычным оконным стеклом, что является его существенным недостатком, так как ультрафиолетовые лучи уничтожают бактерии и оказывают благотворное влияние на человеческий организм. Поэтому большое гигиеническое значение имела бы замена обычного оконного (и электролампового) стекла “увиолевым”, пропускающим ультрафиолетовые лучи. Опыты получения такого стекла уже были проведены, однако выработка его так и не налажена.
Исследования при помощи рентгеновских лучей показали, что стеклообразное состояние вещества (подобно жидкому) отличается от кристаллического неполной упорядоченностью взаимного расположения отдельных элементов пространственной решётки. Поэтому стекло, в противоположность кристаллу, не обладает определённой температурой плавления, а в процессе плавления размягчается постепенно.
В быстро охлаждённом стекле возникают сильные внутренние напряжения, вследствие чего оно легко трескается. Поэтому готовые стеклянные изделия выдерживают в специальных печах, где они подвергаются постепенному охлаждению. Будучи застывшей смесью различных силикатов, стекло при обычных условиях выработки не выделяет их в кристаллическом состоянии и потому остаётся прозрачным. В уже охладившемся стекле процесс кристаллизации идёт настолько медленно, что наблюдать его результаты можно лишь на некоторых старинных изделиях. Однако если нагреть стекло, не доводя его до размягчения, и достаточно долго выдержать при этой температуре, то в результате сильного ускорения процесса кристаллизации происходит выделение кристаллов отдельных силикатов, и стекло становится непрозрачным. Этим иногда пользуются при изготовлении стеклянных изделий, но в большинстве случаев для получения непрозрачных (“молочных”) стёкол специально вводят в расплавленную массу некоторые легко выкристаллизовывающиеся материалы (апатит, криолит и др.).
Путём проводимой в определённых условиях кристаллизации некоторых стёкол могут быть получены материалы, характеризующиеся равномерной тонкозернистой структурой образующихся кристаллов, спаянных друг с другом плёнками незакристаллизованного стекла. Такие ситаллы, сочетающие в себе полезные свойства и кристаллов, и стекла, обладают очень высокой механической прочностью. Интересно, что прочность костей человека обусловлена подобной же двухфазностью их структуры, слагающейся из мелких кристаллов апатита, спаянных органическим веществом (коллагеном).
Ситаллы являются ценным материалом для разнообразных строительных работ. Весьма важно то обстоятельство, что исходным сырьём для их получения могут служить металлургические шлаки.
Получаемая продуванием сквозь жидкую стеклянную массу водяного пара (под давлением) стеклянная вата является прекрасным теплоизоляционным материалом. Этим же качеством, а также звуконепроницаемостью и большой механической прочностью обладает “пеностекло”. Оно образуется при постепенном нагревании до 700—800 °С смеси стеклянного порошка со способными выделять газы веществами и представляет собой как бы напитанную газом стеклянную губку с плотностью 0,2—0,5 г/см3.
Из получаемых на специальных машинах очень тонких (диаметром до долей микрона) стеклянных нитей выделывают ткани, которые используются для ряда технических целей и изготовления спецодежды. Стеклянное волокно обладает высокой удельной прочностью на разрыв, которая тем выше, чем оно тоньше. Даже у “толстых” волокон диаметром 50 мк (что примерно равно диаметру человеческого волоса) она превышает прочность и естественных (хлопок, шерсть), и искусственных (вискоза, капрон) волокнистых материалов. В сочетании с синтетическими полимерами стеклянное волокно даёт легко формуемые при получении материалы (“стеклопластики”), которые примерно в четыре раза легче стали, но могут превосходить её по прочности и практически не подвергаются атмосферной коррозии.
Если стеклянная нить покрыта тончайшим слоем иного стекла с подходяще подобранным показателем преломления, то поступающий в неё с одного конца свет практически без изменения доходит до другого конца. Как бы ни была нить изогнута, закручена и т. д., свет не может из неё “вырваться”, так как постоянно испытывает полное внутреннее отражение. Пучки таких нитей образуют световоды, которые начинают находить использование в самых разнообразных областях. Например, освещая какой-либо труднодоступный объект через часть нитей гибкого световода, можно через другую часть нитей того же световода получить изображение этого объекта. Основанная на свойствах световодов (стеклянных и пластмассовых) волоконная оптика является одним из важных достижений современной техники.
Из стёкол общего типа xNa2O·yB2O3·zSiO2 кислотной обработкой могут быть вымыты окислы щелочного металла и бора, причём образуется пористое стекло, сохраняющее форму исходного. Диаметр пор в таком стекле (состоящем после обработки примерно на 96% из SiО2) может равняться 1500—2000 пм. Подобно цеолитам, оно обладает большой сорбционной активностью.
Сравнительно недавно началось производство кварцевого стекла, представляющего собой по химическому составу почти чистый кремнезём (SiO2). Процесс его выработки в принципе несложен, так как заключается просто в плавлении кварца (обычно — горного хрусталя). Однако поддержание необходимой для этого высокой температуры связано с рядом технических трудностей, обусловливающих высокую стоимость кварцевых изделий.
Плотность кварцевого стекла равна 2,2 г/см3, т. е. она меньше, чем у всех кристаллических модификаций кремнезёма. Выше 200 °С кварцевое стекло начинает заметно пропускать водород и гелий, а выше 1000 °С — и другие газы. Интересной особенностью плавленого кварца является почти полное отсутствие у него упругого последействия, вследствие чего нити и спирали из этого материала незаменимы в производстве ряда точных измерительных приборов.
Наиболее ценным преимуществом кварцевого стекла перед обычным является примерно в 15 раз меньший и почти не изменяющийся с температурой коэффициент термического расширения. Благодаря этому кварцевая посуда переносит без растрескивания весьма резкие изменения температуры: её можно, например, нагреть до красного каления и тотчас опустить в воду. С другой стороны, кварцевое стекло почти вовсе не задерживает ультрафиолетовые лучи и поэтому применяется в аппаратах для их получения. Если плавленый кварц окрасить солями никеля, то получается чёрное стекло, задерживающее все видимые лучи, но пропускающее ультрафиолетовые. Вода и кислоты (кроме HF и Н3РО4) практически не действуют на кварцевое стекло, тогда как щёлочи довольно легко его разъедают. Другим недостатком кварцевого стекла является его большая по сравнению с обычным хрупкость.
Галогениды кремния имеют общую формулу SiГ4 и могут быть получены прямым синтезом по схеме:
Si + 2 Г2 = SiГ4.
Фтор реагирует легко, остальные галогены — лишь при нагревании. Все галогениды SiГ4 бесцветны. При обычных условиях SiF4 газообразен, SiCl4 и SiBr4 представляют собой жидкости, SiI4 — твёрдое тело.
Из химических свойств галогенидов кремния наиболее характерно для них энергичное взаимодействие с водой по схеме:
SiГ4 + 2 Н2О Ы SiO2 + 4 НГ.
Для Cl, Br и I равновесие практически нацело смещено вправо, тогда как для F реакция заметно обратима. Вследствие образования при гидролизе твёрдых частиц SiO2 (точнее, nSiO2•mH2O) пары галогенидов кремния дымят во влажном воздухе.
Некоторые константы галогенидов кремния:
SiF4 SiCl4 SiBr4 SiI4
Теплота образования, кДж/моль ..... . 161 685 460 200
d(SiГ), пм......................... . 155 201 215 243
Температура плавления, °С.......... -90(давл.) -68 +5 122
Температура кипения, °С ........... -95(давл.) +57 153 29
Значительные количества SiF4 получаются как побочный продукт суперфосфатного производства. Фтористый кремний весьма ядовит.
Хлористый кремний получают обычно, действуя хлором на раскаленную смесь SiO2 и угля:
SiO2 + 2 C + 2 Cl2 = 2 CO + SiCl4.
Взаимодействие его при нагревании с оксидами некоторых металлов идёт аналогично реакции с водой (и в отдельных случаях протекает весьма энергично). Например, реакция с Al2O3 идёт по схеме:
3 SiCl4 + 2 Al2O3 = 3 SiO2 + 4 AlCl3 + 100 кДж.
Обмен хлора на кислород происходит также и при взаимодействии SiCl4 c оксидами некоторых неметаллов. Например, четырёххлористый кремний переводит Р2О5 в РОCl3, а SO3 в S2O5Cl2. Интересно, что с металлическим натрием SiCl4 реагирует (с образованием NaCl и выделением аморфного кремния) только при температуре красного каления, тогда как в случаях SiBr4 и SiI4 аналогичная реакция протекает значительно легче. Особым свойством SiI4 является легкая воспламеняемость его нагретых паров на воздухе.
К SiI4 близок по свойствам роданид кремния — Si(NCS)4, который может быть получен взаимодействием SiCl4 и Pb(NCS)2 в бензольном растворе. Вещество это представляет собой бесцветные кристаллы (т. пл. 146, т. кип. 313 °С), под действием воды легко разлагающиеся. Интересно, что в молекуле Si(NCS)4 группировка Si-N=C имеет, по-видимому, линейную структуру, тогда как для молекулы Si(NCO)4 даётся РSiNC = 146° при длинах связей d(SiN) = 169, d(NC) = 121, d(CO) = 116 пм.
Бромистый и йодистый кремний удобнее получать взаимодействием газообразных HВr или HI c парами SiCl4 при нагревании. Если обмен хлора на бром или иод провести не нацело, образуются смешанные галогениды кремния — SiCl3Br, SiCl2Br2, SiClBr3, SiCl3I и др. Взаимодействием SiCl4 и SbF3 при нагревании можно получить аналогичные фторохлориды — SiFCl3, SiF2Cl2, SiF3Cl. При высоких температурах устанавливается некоторое равновесное соотношение между молекулами разного типа. Например, при нагревании SiF2Br2 до 700 °С в смеси одновременно содержатся: SiBr4 (4%), SiFBr3 (25), SiF2Br2 (40), SiF3Br (23), SiF4 (8%). Свойства смешанных производных являются обычно промежуточными по отношению к свойствам соответствующих простых галогенидов.
Кроме простейших галогенидов SiF4, для кремния известны галогенидные производные Si2F6. Общим способом их образования является взаимодействие при высокой температуре паров SiF4 и аморфного Si по обратимой реакции
3 SiF4+ Si Ы 2 Si2F6.
Галогениды Si2F6 представляют собой бесцветные жидкие или твёрдые вещества. Наиболее изученным их представителем является Si2Cl6 (т.пл. 3, т. кип. 147 °С): d(SiSi) = 234 пм, d(SiCl) = 202 пм, РСlSiCl = 110°. Известен аналогичный бромид. Для хлора и брома известны соединения с более длинными цепями атомов кремния вплоть до Si6Cl14 (т.пл. 318 °С) и Si5Br12. Были описаны также продукты состава Si10Cl22 (вязкая жидкость), Si25Cl52 (пластическая масса) и Si10Cl18. Последний имеет бициклическое строение типа Si4Cl82SiClSi4Cl8 и представляет собой жёлтую вазелиноподобную массу.
Все сложные галогениды кремния легко разлагаются водой с образованием в конечном счёте Si(OH)4 и соответствующей галогенводородной кислоты. При проведении реакции на холоду и без избытка воды могут быть выделены неустойчивые промежуточные продукты гидролиза, ещё сохраняющие связи Si-Si в своём составе. Так, Si2Cl6 даёт в этих условиях силикощавелевую кислоту (SiOOH)2, которая выделяется в виде белого порошка, нерастворимого в воде и не обладающего кислыми свойствами. При трении или нагревании она легко разлагается со взрывом, а в щелочах растворяется с выделением водорода. Взаимодействием Si2Cl6 c жидким аммиаком было получено имидное производное состава Si2(NH)3, термическое разложение которого при умеренном нагревании (ниже 500 °С) ведёт к образованию белого рентгеноаморфного нитрида состава SiN.
При нагревании выше 1000 °С порошкообразного кремния в парах SiГ4 протекает обратимая реакция
SiГ4 + Si Ы 2 SiГ2
сопровождающаяся отложением кремния на более холодных частях аппаратуры, т.е. имеющая транспортный характер. Реакция может использоваться для получения чистого кремния.
Для газообразных молекул SiГ2 даются следующие теплоты образования из элементов (кДж/моль): 619 (F), 42 (Br), -79 (I). Кремнийфторид был получен взаимодействием SiF4 с кремнием при 1150 °С и низком давлении (0,1-0,2 мм. рт. ст.). Продолжительности жизни индивидуальных молекул SiF2 [ d(SiF) = 159 пм, РFSiF = 101°] гораздо выше, чем у молекул галогенкарбенов. При их полимеризации образуется белая или желтоватая каучукоподобная масса (SiF2)n, нерастворимая в органических растворителях и разлагаемая водой. Из продуктов её термического разложения при 300 °С, помимо Si2F6 были выделены Si3F8 (т. пл. -1, т. кип. 42 °С), Si4F10 (т. пл. 67, т. кип. 85 °С) и другие члены вплоть до Si14F30. На воздухе они самовоспламеняются.
Жёлто-коричневый (SiBr)n может быть получен восстановлением растворённого в эфире SiBr4 магнием, а аналогичный по составу (SiCl)n — взаимодействием силицида магния с ICl. Оба моногалогенида кремния нерастворимы в бензоле, а водой разлагаются. Структура их отвечает “паркету” из шестичленных колец. Были описаны также (SiF)n и (SiI)n.
Выше 380 °С (SiCl)n начинает отщеплять летучие хлориды кремния, постепенно приобретая красную окраску, а выше 550 °С превращается в почти не содержащую хлора тёмно-зелёную массу, обладающую металлическим блеском и рентгеноаморфную. Кристаллизация её наступает лишь при выдерживании выше 800 °С.
Известны и некоторые производные, отвечающие кислотной функции Si(OH)4. Лучше других изучен кремнийтетрацетат — Si(CH3COO)4, представляющий собой бесцветное кристаллическое вещество (т. пл. 110 °С), разлагающееся при нагревании выше 160 °С, растворимое в ацетоне и бензоле, но тотчас подвергающееся гидролизу под действием воды.
Белый кристаллический кремнийтетразид — Si(N3)4 — весьма взрывчат. Нитрат и перхлорат кремния в индивидуальном состоянии не получены, но были выделены в виде взрывчатых двойных соединений состава Si(NO3)4·2C5H5N и Si(ClO4)4·2CH3CN.
Из фосфатов кремния наиболее изучено производное состава SiO2·P2O5, известное в двух формах — низкотемпературной (ниже 1000 °С) и высокотемпературной (т. пл. 1290 °С). Последняя не разлагается не только водой, но и плавиковой кислотой. С химической точки зрения вещество такого состава может быть или метафосфатом силицила — SiO(PO3)2, или пирофосфатом кремния — SiP2O7. Возможно, что одна из этих формул отвечает высокотемпературной форме, а другая — низкотемпературной. Был получен также нормальный ортофосфат кремния – Si3(PO4)4. Он обладает значительной твёрдостью и не разлагается кислотами (в том числе и плавиковой).
При взаимодействии SiF4 с плавиковой кислотой образуется комплексная фторокремневая (иначе кремнефтористоводородная) кислота:
2 HF + SiF4 = H2[SiF6].
Безводная Н2SiF6 не существует. Напротив, в водном растворе она устойчива и является сильной двухосновной кислотой.
Для реакции образования Н2SiF6 в разбавленных водных растворах К= =[SiF4][F’]2/[SiF6”] = 7·10-7. Её 13,3%-ный раствор перегоняется без разложения, а охлаждением крепких водных растворов она может быть выделена в виде малоустойчивых кристаллогидратов. Кислотные свойства Н2SiF6 выражены сильнее, чем у серной кислоты, — её степень диссоциации в 0,1 н. растворе составляет около 75%. Аналогичные H2SiF6 комплексные кислоты других галогенов не образуются. Особое положение фтора связано, по-видимому, со значительно меньшим объёмом F- по сравнению с Cl-, Br- и I-. Ядерное расстояние Si-F в ионе SiF62- равно 169 пм, а эффективный радиус этого иона оценивается в 240 пм.
Большинство солей этой кислоты (кремнефтористых или фторосиликатов) бесцветно и хорошо растворимо в воде. Из производных обычных металлов наиболее труднорастворимы соли калия и особенно бария.
При накаливании фторосиликаты разлагаются на SiF4 и соответствующий фтористый металл. Так, термическая диссоциация по схеме:
Na2SiF6Ы 2 NaF + SiF4
становится заметной примерно с 450 °С. Интересно, что термический распад K2SiF6 идёт, по-видимому, с промежуточным образованием K3SiF7. Фторосиликаты Ca, Sr и Ba разлагаются на SiF4 и MF2 соответственно при 370, 420 и 560 °С.
Аналогично термическому протекает распад фторосиликатов и при растворении их в жидком HF. Реакция идёт по схеме:
Na2SiF6 + 2 HF = 2 NaHF2 + SiF4.
Аммиаком фторосиликаты разрушаются с выделением свободной кремневой кислоты по схеме:
Na2SiF6 + 4 NH4OH = 2 NaF + 4 NH4F + Si(OH)4.
Подобным же образом идёт процесс и под действием сильных щелочей (NaOH или КОН), но в этом случае при избытке щёлочи образуется силикат соответствующего металла, и поэтому осадок кремневой кислоты не выпадает.
Вследствие образования H2SiF6 приводившаяся ранее общая схема гидролиза галогенидов кремния для SiF4 является несколько более сложной и выражается уравнением:
3SiF4 + 2 H2O = SiO2 + 2 H2SiF6.
Таким путём обычно и получают кремнефтористоводородную кислоту. Образование H2SiF6 всегда имеет место также при взаимодействии раствора HF с SiO2 или стеклом.
Свободная H2SiF6 используется для фторирования воды и в пивоварении (как дезинфицирующее средство), а малорастворимые фторосиликаты Na (0,7 вес. %) и Ва (0,01 %) — для борьбы с вредителями сельского хозяйства. Малой растворимостью K2SiF6 (0,2%) иногда пользуются для получения свободных кислот (например, HClO3) исходя из их калийных солей. Легкорастворимые фторосиликаты Mg, Zn и Al под техническим названием “флюаты” находят применение в строительном деле (для придания водонепроницаемости цементированным поверхности).
Водородные соединения кремния (кремневодороды илисиланы) получаются в смеси друг с другом и с водородом при действии разбавленной НСl на силицид магния (Mg2Si). По своему составу и структурным формулам кремневодороды (SiH4, Si2H6 и т. д. вплоть до последнего известного члена — Si8H18) аналогичны углеводородам ряда метана. Большое сходство наблюдается и в отношении физических свойств: подобно углеводородам, силаны бесцветны, первые члены гомологического ряда при обычных условиях газообразны, следующие представляют собой жидкости. Напротив, общая химическая характеристика обоих классов соединений различна: в противоположность инертным углеводородам силаны весьма реакционноспособны. В частности, на воздухе они легко воспламеняются и сгорают до SiO2 и воды, причём горение сопровождается очень большим выделением тепла (1425 кДж/моль SiH4).
Устойчивость силанов уменьшается по мере увеличения числа атомов кремния в молекуле. В том же направлении уменьшаются и их относительные количества, получающиеся при взаимодействии Mg2Si с кислотой или раствором NH4Br в жидком аммиаке (последний способ даёт значительно лучший выход силанов). Путём сильного охлаждения газовой смеси и её фракционной перегонки в отсутствие воздуха были выделены отдельные кремневодороды вплоть до Si8H18. Некоторые константы первых членов ряда приводятся ниже:
SiH4 Si2H6 Si3H8 Si4H10
Температура плавления, °С....... –185–132 –117 –86
Температура кипения, °С......... –112 –14 +53 107
Тетрасилан был получен в двух изомерных формах (аналогичных нормальному бутану и изобутану).
Все силаны обладают характерным запахом и сильно ядовиты. При нагревании высшие члены ряда распадаются с образованием (SiH)x, SiH4 и H2. Окислителями силаны переводятся в H2O и SiO2. Со свободными галогенами они реагируют аналогично углеводородам, последовательно обменивания на галоген один атом водорода за другим. С галогеноводородами (например, HCl) в присутствии катализатора (AlCl3) идёт при нагревании подобная же (но не имеющая аналогии в химии углерода) реакция обмена водорода на галоген по схеме:
SiH4+ HCl = H2 + SiH3Cl.
C концентрированной H2SO4 силаны (подобно углеводородам) не реагируют. Они хорошо растворимы в органических растворителях, но почти нерастворимы в воде. Последняя разлагает их по схеме:
SiH4 + 2 H2O = 4 H2 + SiO2.
Однако с тщательно очищенной водой в кварцевых сосудах реакция идёт настолько медленно, что остаётся практически незаметной. В присутствии следов кислот, и особенно щелочей, она значительно ускоряется, и при обычных условиях вода разлагает за сутки уже около 20% исходного количества SiH4.
Cилан является эндотермичным соединением (теплота его образования из элементов равна –33 кДж/моль). До 450 °С он термически устойчив, а при дальнейшем нагревании начинает постепенно разлагаться на элементы. Молекула SiH4 представляет собой правильный тетраэдр с атомом кремния в центре [d(SiH4) = 148 пм].
В молекуле дисилана d(SiH) = 149, d(SiSi) = 233 пм. Термический распад Si2H6 начинается уже выше 300 °С. Интересно, что дисилан реагирует с CСl4, тогда как силан с ним не взаимодействует. Это указывает, по-видимому, на неполную экранированность кремния в дисилане.
Непредельным углеводородам формально аналогичны гидриды кремния типов (SiH2)n и (SiH)n. Полисилен — (SiH2)n — образуется при растворении СаSi безводной уксусной кислотой. Он представляет собой светло-коричневое твёрдое вещество, нерастворимое в обычных растворителях и самовоспламеняющееся на воздухе. При нагревании до 380 °С (SiH2)n разлагается с выделением кремния и смеси летучих силанов, а водой по схеме:
SiH2 + 2 H2O = SiO2 + 3 H2.
Более или менее близкие по составу (SiH)n образуются при термическом распаде летучих силанов. По свойствам они промежуточны между (SiH2)n и элементарным кремнием. Синтетически (SiH)n может быть получен по схеме:
2 SiHBr3 + 3 Mg = 3 MgBr2 + 2 SiH
(реакция проводится в абсолютном эфире при полном исключении кислорода). Получаемый (SiH)n представляет собой жёлтое хрупкое рентгеноаморфное вещество.
Путём гидролиза частично галогенозамещённых кремневодородов был получен ряд производных кремния, называемых силоксанами и характеризующихся наличием в молекуле группировок Si–O–Si. Простейшим их представителем является (SiH3)2O, который может быть получен по схеме:
2 SiH3Г + Н2О = 2 НГ + (SiH3)2O
Аналогичные простым эфирам силоксаны представляют собой газообразные или жидкие вещества, быстро разлагаемые щелочами и медленно (с образованием промежуточных продуктов) — водой.
Примером более сложного силоксана может служить продукт гидролиза силикохлороформа по схеме:
2 НSiCl3 + 6 H2O = 6 HCl + 2 HSi(OH)3 = 3 H2O + 6 HCl + H2Si2O3.
Полисилоксан (Н2Si2O3)n представляет собой белое твёрдое вещество, структура которого слагается из соединенных связями Si–Si колец, образованных группами SiH и атомами кислорода. При нагревании до 500 °С он медленно отщепляет водород, давая в конечном счёте оксид кремния простейшей формулы Si2O3.
Для кремния известно большое число различных кремнийорганических соединений. В качестве простейшего их представителя можно рассматривать силилметил — H3SiCH3, который представляет собой бесцветный газ (т. кип. –57 °С). В отличие от этана и дисилана, молекула его полярна (m = 0,73). Некоторые кремнийорганические соединения во многом аналогичны соответствующим производным углерода. Например, этиловые эфиры ортокремневой и ортоугольной кислот [Si(OC2H5)4 и C(OC2H5)4] представляют собой бесцветные жидкости, кипящие соответственно при 166 и 158 °С. Кипячением первого из этих соединений с водой может быть получен коллоидный раствор SiO2, свободный от электролитов.
Кремнийорганические соединения, как правило, устойчивы на воздухе и нерастворимы в воде. Синтез высокомолекулярных производных со скелетом из группировок —Si—O—Si—O— (т. н. силиконов) открыл возможность их широкого практического использования.
Свойства построенных по типу
R R R R R R
Ѕ Ѕ Ѕ Ѕ Ѕ Ѕ
—Si—O—Si—O—Si—O— или —Si—O—Si—O—Si—O—
Ѕ Ѕ Ѕ Ѕ Ѕ Ѕ
R R R R O R
Ѕ
полимерных молекул определяются в основном отношением числа органических радикалов (R) к числу атомов кремния: при R:Si > 2 получаются вязкие жидкости, при R:Si » 2 — эластичные каучукоподобные массы, а при R:Si > 2 используются, например, как смазки (почти не изменяющие свою вязкость в широком интервале температур), с R:Si » 2 — при изготовлении резин специального назначения (в частности, термо- и морозостойких), с R:Si
Ниже сопоставлены средние длины и энергии связей в соединениях кремния:
Связь ............ Si–Si Si–H Si–O Si–S Si–N Si–C Si–F Si–Cl Si–Br Si–I
Длина, пм ........ 234 148 164 215 174 187 158 203 217 244
Энергия, кДж/моль 220 345 450 265 320 315 565 410 310 235
Следует отметить, что литературные данные по энергиям рассматриваемых связей иногда сильно расходятся.
Приведённые выше средние длины связей Si–Э, как правило, меньше сумм соответствующих ковалентных радиусов. Обусловлено это, по-видимому, полярным характером рассматриваемых связей. Зависимость сокращения ядерных расстояний от расчётных полярностей связей имеет довольно закономерный характер.
Существенное отличие химии кремния от химии углерода обусловлено прежде всего относительно малой прочностью связей Si–Si. Поэтому цепочки из атомов Si разрываются гораздо легче, чем углеродные, особенно если имеется возможность образования наиболее характерной для кремния связи с кислородом. Прямым следствием является резкое уменьшение числа устойчивых кремниевых соединений по сравнению с углеродными. Последнее в свою очередь косвенно отражается на сравнительном многообразии органического и минерального мира: в то время как отдельных видов живых организмов описано более миллиона, различных минералов известно около трёх тысяч.
Большая устойчивость связи Si–O налагает свой отпечаток на всю химию кремния. В противоположность углероду, для которого единственной гидратной формой высшего окисла является угольная кислота, для кремния известен ряд производных от самых различных гидратов кремнезёма. Само многообразие этих производных — природных силикатов — обусловлено именно прочностью связи Si–O–Si, играющей в химии кремния такую же основную роль, как связь С–С в органической химии. На современном этапе своего развития неорганическая химия кремния находится ещё в зачаточном состоянии. Впоследствии она, вероятно, выделится в отдельную область, настолько же важную для геологии, насколько важна органическая химия для биологии.
Общее направление круговорота С и Si в природе прямо противоположно: для углерода характерно постепенное связывание СО2 с образованием солей Н2СО3, для кремния — разрушение солей кремневых кислот (природных силикатов) с выделением свободного кремнезёма.
ХIII. Первая группа периодической системы
Подгруппа меди. По распространенности в природе элементы этой подгруппы стоят далеко позади соответствующих щелочных металлов. Если содержание самой меди в земной коре оценивается еще довольно большой величиной — 0,003%, то доля серебра составляет уже только 2Ч10-6 %, а золота — 5Ч10-8 %.
Природная медь слагается из изотопов 63Сu (69,1 %) и 65Сu (30,9 %), серебро — из изотопов 107Аg (51,35 %) и 109Аg (48,65 %), тогда как золото является «чистым» элементом (197Аu).
В основном состоянии элементы подгруппы меди имеют строение внешних электронных оболочек 3d104s1 (Сu), 4d105s1 (Аg), 5d106s1 (Аu) и одновалентны. Возбуждение ближайших потенциально трехвалентных состояний Сu (3d94s14р1), Аg (4d95s15р1) и Аu (5d96s16р1) требует затраты соответственно 464, 673 и 502 кДж/моль.
М е д ь принадлежит к интересным в биологическом отношении элементам. Она является катализатором внутриклеточных окислительных процессов.
Установлено, что небольшие количества меди необходимы для нормального развития растений и удобрение почв (особенно — болотистых и песчаных) ее соединениями часто сопровождается резким повышением урожайности. По отношению к избыточному содержанию меди устойчивость растительных организмов очень различна.
Из животных организмов больше всего содержат меди некоторые моллюски (осьминоги, устрицы). У высших животных она накапливается главным образом в печени и клеточных ядрах других тканей. Недостаточное поступление Си в организм (ежедневная норма для человека составляет около 5 мг) ведет к уменьшению новообразования гемоглобина и развитию анемии, которая может быть излечена введением соединений меди в пищу. Из отдельных видов последней наиболее богаты медью молоко и дрожжи. Интересно, что в крови беременных найдено удвоенное по сравнению с нормальным содержание меди.
Следы с е р е 6 р а (порядка 0,02 мг Аg на 100 г сухого вещества) содержатся в организмах всех млекопитающих, но его биологическая роль не ясна. У человека повешенным содержанием Аg (0,03 мг на 100 г свежей ткани, или 0,002 вес.% в золе) характеризуется головной мозг. Интересно, что в изолированных ядрах его нервных клеток — нейронов (число которых у человека составляет около 15 млрд.) — серебра гораздо больше (0,08 вес.% в золе). С пищевым рационом человек получает в среднем около 3,1 мг Ag за сутки. Относительно много его содержит яичный желток (0,2 мг в 100 г). Выводится серебро из организма главным образом с калом.
Относительно содержания в организмах и биологической роли з о л о т а пока ничего определённого не известно. Отмечалось наличие его в зернах, листьях и стеблях кукурузы. Воды океана содержат переменные количества золота, (от ничтожных следов до 65 мг/т).
Медь и серебро встречаются главным образом в виде сернистых соединений и чаще всего совместно с сернистыми рудами других металлов. Из отдельных минералов меди наиболее важны х а л ь к о п и р и т (СuFеS2) и х а л ь к о з и н (Си2S). Гораздо меньшее промышленное значение имеют кислородсодержащие минералы — к у п р и т (Сu2О), м а л а х и т [(СuОН)2СО3] и др.
Сернистое серебро как отдельный минерал (а р г е н т и т — Аg2S) встречается сравнительно редко. Напротив, весьма обычно нахождение Ag2S в виде примеси к сернистым рудам Рb, Zn и Сu.
Из природных соединений золота с другими элементами наиболее характерны теллуристые минералы — к а л а в е р и т (АuТe2) и др. Однако гораздо более обычной формой природного нахождения золота является самородное состояние в виде вкраплений в горных породах, россыпей золотого песка и отдельных самородков, иногда значительной массы. Одной из наиболее богатых месторождениями золота стран является Россия.
Подобно золоту, иногда встречаются в самородном состоянии также серебро и медь. Вероятно, именно поэтому все три элемента и были известны человечеству еще в самой глубокой древности. Из них медь являлась первым металлом, широко использованным для изготовления орудий труда.
Самые крупные когда-либо найденные самородки золота, серебра и меди весили соответственно 112 кг, 13,5 т и 420 т, Интересно, что у некоторых крупных самородков меди все выступающие части оказались отбитыми каменными топорами.
Наиболее богатой з о л о т о м страной древнего мира был Египет (максимум добычи — до 50 т ежегодно — приходился, по-видимому, на время около 1500 лет до н. э.). Из европейских стран главным поставщиком золота являлся в прошлом Пиренейский полуостров.
В древности и даже в прошлом столетии добыча золота производилась почти исключительно из россыпей, образовавшихся путем выветривания и размыва природными водами содержащих Аu горных пород. Значительно больше золота добывается в настоящее время непосредственно из таких пород, которые предварительно подвергаются дроблению и размалыванию.
При среднем содержании золота эксплуатируемых ныне месторождений всего лишь в 0,001 % отделение его от пустой породы старым, основанным на различии плотностей, способом отмывки водой становится неэкономичным. Вместо него пользуются двумя другими способами: ртутным и цианидным.
Первый из них основан на образовании амальгамы Аu при обработке размолотой с водой породы металлической ртутью. Сама по себе растворимость золота в ртути невелика: при обычных условиях она составляет лишь 0,13 ат. % (а серебра и меди соответственно 0,07 и 0,006 ат. %). Однако, помимо растворенного Аu, амальгама содержит также соединения обоих металлов (известны АuНg2, Аu2Hg и Au3Hg и, главное, суспензию захваченных целиком (за счет смачивания) крупинок золота.
Полученную амальгаму продавливают сквозь замшу и жидкую часть используют вновь (так как она лучше смачивает золото, чем чистая ртуть), а твердую подвергают перегонке, причем ртуть отгоняется, а золото остается и перегонном аппарате. Важнейшим недостатком ртутного способа является неполнота выделения золота, так как мельчайшие его частички плохо смачиваются ртутью и поэтому не амальгамируются.
В отличие от ртутного цианидный способ позволяет практически полностью выделять золото даже из самых бедных пород. Для извлечения Аu размолотую золото-содержащую породу обрабатывают при доступе воздуха очень разбавленным (0,03-0,2 %) раствором NаСN. При этом золото по уравнению 4 Аu + 8 NaCN + 2 H2O + O2 = 4 Nа[Аu(СN)2] + 4 NаОН переходит в раствор, из которого затем выделяется действием металлического цинка:
2 Na[Аu(СN)2]+ Zn = Nа2[Zn((CN)4]+ 2 Au.
Очистка полученного тем или иным путем золота от примесей (“аффинаж”) производится чаще всего обработкой его горячей концентрированной Н2SО4, или электролитически.
Большая часть добываемого в настоящее время с е р е б р а получается при переработке не собственно серебряных руд, а содержащих примеси Ag сернистых руд Рb, Zn и Сu. Особенно характерно для серебра совместное нахождение со свинцом. Состав часто встречающихся серебро-свинцово-цинковых руд колеблется в зависимости от месторождения. Например, руда может содержать 15-20 % Zn, 10-15 % Pb и 0,15-0,25 % Аg.
Взаимная растворимость расплавленных Zn и Рb c понижением температуры быстро уменьшается. Поэтому полученный восстановлением серебро-свинцово-цинковых руд жидкий металл при охлаждении разделяется на два слоя нижнем — свинцовом — остается очень мало цинка, в верхнем — цинковом — мало свинца. Так как растворимость серебра в цинке значительно больше, чем в свинце, почти все имевшееся в первоначальном сплаве Аg остается при этом в цинковом слое, из которого затем и выделяют путем отгонки цинка (и окисления свинца). Из руд бедных другими металлами Аg часто получают путем обработки их раствором NaCN при доступе воздуха. При этом серебро (аналогично золоту) переходит в раствор, из которого может быть выделено действием металлического цинка.
Окончательную очистку Аg производят чаще всего путем электролиза, причем электролитом служит раствор АgNО3, а анодом — пластина, отлитая из сырого серебра. При пропускании тока на катоде осаждается чистое серебро, а содержавшиеся в сыром металле примеси Сu, Рb и Zn удерживаются в растворе. Обычно также содержащееся в виде примеси Ag остается нерастворенным и собирается на дне сосуда у анода.
10) Выплавка м е д и из ее сернистых руд слагается из нескольких стадий. Прежде всего руду обжигают на воздухе для удаления главной массы содержащейся в ней серы. Операцию эту проводят в специальных механических печах или в кипящем слое. Обожженную руду с добавкой флюсов переплавляют затем в отражательных печах (рис. Х111-37). При этом пустая порода и часть железа переходят в шлак, а Сu2S, FеS и небольшие количества других примесей сплавляются в «штейн» (который собирается на дне печи). Последний переводят в специальные конверторы, где медь освобождают от серы и железа путем продувания сквозь расплавленную массу воздуха. Так как выгорание серы и железа сопровождается выделением большого количества тепла, затрат топлива для проведения этого процесса не требуется.
Получаемая описанным способом черновая медь содержит обычно 95-98 % Сu. От большей части имеющихся в ней примесей она может быть освобождена переплавкой на поду отражательной печи, причем получается «штыковая медь» с содержанием до 99,7 %. Перепллвка меди для ее очистки была описана уже в книге Бирингуччио.
Так как очистка (“рафинирование”) меди электролизом обходится дороже, ею пользуются лишь тогда, когда необходимо получить особенно чистый металл. Электролитом при этом обычно служит раствор СuSO4. Содержавшиеся в виде примесей к меди Fе и Zn при электролизе остаются в растворе, Аg и Аu (а также Рt) оседают на дно сосуда, (стоимость получаемых таким путем драгоценных металлов играет важную роль для покрытия издержек по электролизу.
11) В качестве побочного продукта при выплавке меди получается большое количество сернистого газа. Поэтому весьма рациональным является комбинирование медеплавильных заводов с заводами для выработки серной кислоты. Подобные сочетающие в себе металлургию и химию комбинаты позволяют наладить к о м п л е к с н о е и с п о л ь з о в а н и е с ы р ь я с извлечением из него всех ценных составных частей.
Например при полном комплексном использовании медных колчеданов наряду с медью могут быть получены большие количества железа и серной кислоты, а, кроме того, мышьяк, цинк, свинец, сурьма, висмут, серебро и золото (из примесей исходного колчедана).
12) Выплавка меди из оксидных руд несравненно проще, чем из сернистых, так как сводится в основном к легко протекающему восстановлению углем. Именно таким путем и добывали, по-видимому, медь в древности. Выработка ее в Египте достигала значительных размеров уже за 3000 лет до н. э.
13) Так как содержание Сu в эксплуатируемых рудах обычно не превышает 1 %, для рентабельной их переработки необходимо предварительное обогащение, т. е. повышение процентного содержания металла в руде за счет отделения его соединений от пустой породы. Это достигается применением метода флотации руд, основанного на различных адсорбционных свойствах поверхностей частиц сернистых металлов и окружающей их пустой породы силикатного типа.
Если в водный раствор какого-либо малополярного органического вещества поместить смесь порошков силиката и сернистого металла, то на поверхности первого будут адсор6ироваться почти исключительно молекулы воды, а на поверхности второго — малополярные молекулы растворенного органического вещества. С другой стороны, если сквозь такой раствор пропускать пузырьки воздуха, то последние будут адсорбировать на своей поверхности также почти исключительно малополярные молекулы.
Покрытые о д и н а к о в ы м и адсорбированными слоями частицы сернистых металлов и продуваемые сквозь раствор пузырьки воздуха легко слипаются друг с другом. Напротив, частицы силикатов к пузырькам воздуха не прилипают. Если в воде, содержащей небольшую примесь малополярного органического вещества (например, соснового масла), взболтать порошок тонко измельченной медной руды и сквозь всю систему продувать воздух, то частицы сернистой меди будут вместе с воздушными пузырьками подниматься вверх и перетекать через край сосуда в виде пены, а частицы силикатов осядут на дно.
На этом и основан флотационный метод обогащения, при помощи которого ежегодно перерабатывается более 100 млн. т сернистых руд различных металлов. Обогащенная руда — концентрат — содержит обычно 20 ё 30 % меди Флотация дает также возможность разработки более бедных месторождений, которые без ее помощи вообще не могли бы быть освоены. Вместе с тем методика проведения флотационных процессов настолько разработана, что при помощи так называемой с е л е к т и и н о й (избирательной) флотации удается не только отделять руду от пустой породы, но во многих случаях успешно разделять отдельные минералы полиметаллических руд.
14) Наряду с обычным получением меди путем процессов, проводимых при высоких температурах (п и р о м е т а л л у р г и я), большое значение имеют методы ее извлечения, основанные на обработке руд теми или иными жидкостями (г и д р о м е т а л л у р г и я). Для извлечения меди из особенно пригодных для гидрометаллургической переработки окисленных руд часто пользуются разбавленным раствором серной кислоты. Медь сульфидных руд может быть переведена в раствор по схеме:
Сu2S + 2 Fе2(SO4)3 = 4 FеSO4 + 2 СuSO4 + S
Из образующегося разбавленного раствора медной соли металл вылепляют затем либо электролизом, либо действием металлического железа. Окисление сернистых медных руд может быть сильно ускорено с помощью серобактерий.
В настоящее время на долю гидрометаллургии приходится около 15 % всей мировой добычи меди. Широкие перспективы дальнейшего развития открывает перед этим методом принципиальная возможность его осуществления подземным способом, в известной мере аналогичным подземной газификации угля.
В элементарном состоянии Сu, Аg и Аu представляют собой металлы соответственно красного, белого и желтого цвета.
Все три металла характеризуются значительными плотностями, довольно высокими температурами плавления и сравнительно малой твердостью. Их тягучесть и ковкость исключительно велики. Из любого металла можно вытянуть проволоку диаметром в 0,001 мм (которая примерно в 50 раз тоньше человеческого волоса), а путем ковки или прокатки Au получают листочки («золотую фольгу») толщиной до 100 нм. Они имеют в отраженном свете желтый, а в проходящем — зеленый цвет. По электро- и теплопроводности элементы подгруппы меди превосходят все остальные металлы.
Их важнейшие константы сопоставлены в приводимой таблице:
| Сu | Ag | Au | |
Плотность, г/см3 | 9,0 | 10,5 | 19,3 |
| Твердость (алмаз = 10) | 3,0 | 2,7 | 2,5 |
| Электропроводность (Hg = 1) | 57 | 59 | 40 |
| Теплопроводность (Hg = 1) | 51 | 57 | 40 |
Температура плавления, °С | 1085 | 962 | 1064 |
Температура кипения, °С | 2880 | 2160 | 2850 |
15-21
Друг с другом и со многими другими металлами Сu, Аg и Аu образуют сплавы. В частности, все они сплавляются со ртутью (труднее остальных Сu). На легком образовании амальгамы основан иногда применяемый метод извлечения золота из содержащих его горных пород.
22-25
Химическая активность меди и ее аналогов невелика и по ряду Сu-Аg-Аu быстро уменьшается. Золото и серебро на воздухе не изменяются, а медь постепенно покрывается плотной зеленовато-серой пленкой основных углекислых солей. С кислородом под обычным давлением непосредственно соединяется только медь (при нагревании), с серой — уже не только Сu, но и Аg. С водородом, азотом и углеродом Сu, Аg и Аu не реагируют даже при высоких температурах.
26-29
Значительно легче, чем с другими элементами, идет взаимодействие меди и ее аналогов со свободными хлором, бромом и иодом. Медь и серебро медленно соединяются с ними уже при обычной температуре. Золото подобным же образом относится к брому, но с сухим хлором и иодом реагирует только при нагревании. В водном растворе хлора («хлорной воде») золото растворяется легко. При этих условиях реакция взаимодействия медленнее всего протекает у серебра ввиду образования на его поверхности слоя труднорастворимого AgCl.
30
В ряду напряжений все три элемента располагаются п р а в е е водорода, причем медь стоит почти рядом с ним, а золото — дальше всех остальных металлов. Поэтому в растворах таких кислот, как НСl, Н2SO4 и т. п., при отсутствии окислителей не растворяется даже медь. В кислотах, одновременно являющихся окислителями (НNО3, горячая концентрированная Н2SO4 и т. п.), медь и серебро растворяются легко, а золото лишь в том случае, когда окислительные свойства кислоты выражены особенно сильно (например, золото растворяется в горячей безводной Н2SеO4). Хорошо растворяет его и царская водка, что было известно еще алхимикам. Лучшим растворителем Аu является насыщенная хлором соляная кислота, взаимодействующая с золотом по уравнению
2 Аu + 3 Сl2 + 2 НСl = 2 Н[АuСl4]
По отношению к сильным щелочам элементы подгруппы меди устойчивы.
31-32
В своих соединениях серебро главным образом одновалентно. Напротив, Сu и Au образуют по два довольно хорошо изученных ряда производных: Сu — одно- и двухвалентного, Аu — одно- и трехвалентного элемента. Более устойчивы и практически важны в большинстве случаев производные двухвалентной меди и трехвалентного золота. Растворимые соединения Сu, Аg и Аu ядовиты.
Наиболее характерной особенностью большинства соединений рассматриваемых элементов является л е г к о с т ь в о с с т а н о в л е н и я их до металлов. В соответствии с положением в ряду напряжений легче всего восстанавливаются производные Аu, труднее всего — Cu. Другой весьма характерной чертой является их с к л о н н о с т ь к к о м п л е к с о о б р а з о в а н и ю.
33-42
Оксиды типа Э2О окрашены в характерные цвета: Сu2О — красный, Аg2O — темно-бурый, Аu2О — серо-фиолетовый. В воде монооксиды почти не растворимы и присоединяют ее с образованием гидроксидов ЭОН лишь незначительно. Наоборот образующиеся при действии щелочей на соли одновалентных Сu, Аg и Аu осадки гидроксидов легко отщепляют воду, частично переходя в соответствующие монооксиды.
Гидроксиды ЭОН являются основаниями средней силы. Так, влажный Аg2О отчетливо окрашивает лакмусовую бумажку в синий цвет. С другой стороны, все рассматриваемые монооксиды несколько растворимы в крепких растворах щелочей, что указывает на наличие у них признаков амфотерности. Однако кислотная функция ЭОН выражена несравненно слабее основной и соответствующие ей соли не выделены.
43-49
В противоположность производным щелочных металлов многие соли с катионами Сu+, Аg+ и Аu+ окрашены даже при наличии в молекуле бесцветного аниона. Особенно это относится к производным Аu+ которых характерен желтый цвет. В воде соли Сu+ и Аu+ почти нерастворимы, причем во влажном состоянии и те и другие неустойчивы. Ввиду этого иметь с ними дело приходится редко.
50
Напротив, соединения Аg+ находят разнообразное применение. Хотя большинство солей Аg+ малорастворимо в воде, однако для серебра известен и ряд хорошо растворимых. В водном растворе они обычно показывают нейтральную реакцию на лакмус. Наиболее практически важно а з о т н о к и с л о е серебро, которое получают по реакции
3 Аg + 4 НNО3 = 3 АgNО3 + NО + 2 H2O
Эта хорошо растворимая в воде бесцветная соль служит одним из самых употребительных реактивов и обычным исходным продуктом для приготовления остальных соединений серебра. Из последних большое значение имеют почти нерастворимые в воде галоидные соли — белое АgСl, желтоватое АgВr и желтое АgI, так как идущий под действием света распад их с выделением металлического Аg лежит в основе фотографического процесса. Подобно галогенидным солям, постепенно распадается под действием света и большинство других соединений серебра. Поэтому их (а также их растворы) хранят обычно в банках из темного стекла.
С рядом молекул и ионов (NН3, СN-, S2O32- и др.) соли одновалентен Сu, Аg и Au образуют комплексные соединения. Так как последние большей частью хорошо растворимы, их образование влечет за собой растворение многих нерастворимых в воде исходных солей. Например, вследствие комплексообразования по схеме
АgСl + 2 NН3 = [Аg(ХН3)2]Сl
нерастворимое в воде AgСl растворяется в NН4ОН. У производных Сu+ и Ag+ комплексообразование часто сопровождается не только их растворением, но и повышением устойчивости.
51-119
Из солей, в которых элементы подгруппы меди д в у х в а л е н т н ы, хорошо изучены и имеют практическое значение только производные самой меди. Ее черный оксид (СuО) иногда встречается в природе и легко может быть получен прокаливанием Сu на воздухе. В воде она нерастворима, а в кислотах растворяется, образуя соответствующие соли.
Отвечающий оксиду меди г и д р о к с и д Сu(OH)2; выделяется в виде голубого осадка при действии избытка щелочи на растворы солей Сu2+. В воде она почти нерастворима, а при нагревании легко переходит в СuО. Переход этот происходит даже при кипячении содержащей Сu(ОН)2 жидкости.
Для гидроксида меди характерны довольно слабо выраженные основные свойства. С кислотами он дает соли, большинство которых образует кристаллогидраты и хорошо растворимо в воде. В достаточно разбавленных растворах цвет всех солей двухвалентной меди с бесцветными анионами сине-голубой (цвет полностью гидратированного иона Сu••). Напротив, окраска твердых солей Сu2+ различна. Напротив практически важная из них — м е д н ы й к у п о р о с (CuSO4·5H2O) — имеет синий цвет.
Комплексообразание для двухвалентной меди весьма характерно. С соответствующими солями щелочных металлов соли Сu2+ дают двойные соединения содержащие медь в составе комплексных анионов (например, [СuСl4]2-). Однако большинство последних неустойчиво и в растворе распадается на свои составные части. Значительно устойчивее очень характерный для двухвалентной меди синий комплексный катион [Cu(NН3)4]••, образующийся при прибавлении избытка аммиака к растворам солей Cu2+ по реакции, например:
СuSO4 + 4 NН4OН = [Сu(NН3)4]SO4 + 4 Н2О
В связи с интенсивной окраской этого комплекса аммиаком можно пользоваться как реактивом на медь.
120-157
Из солей т р е х в а л е н т н ы х элементов подгруппы меди хорошо изучены только производные Аu. Обычным исходным продуктом для их получения служит коричнево-красное хлорное золото (АuСl3), образующие около 200 °С при действии избытка хлора на порошок Аu.
О к с и д золота (Аu2О3) может быть получен только косвенным путем. Он представляет собой нерастворимый в воде коричневый порошок, легко отщепляющий кислород при нагревании.
Красно-бурый г и д р о к с и д Au(ОН)3 выпадает в осадок при действии сильных щелочей на крепкий раствор АuСl3. Он амфотерен, причем кислотная функция выражена сильнее основной.
Соли Au(ОН)3 с основаниями — а у р а т ы — образуются при его растворении в сильных щелочах и производятся от комплексной кислоты Н[Аu(ОН)4]. Соли, отвечающие основной функции Аu(ОH)3, могут быть получены его растворением в сильных кислотах. Большинство производных трехвалентного золота окрашено, чаще всего в цвета желтых оттенков.
Характерной особенностью Аu3+ является склонность к образованию комплексных анионов. Например, при взаимодействии АuСl3 с водой по схеме
Н2О + АuСl3 = Н2[ОАuСl3]
получается коричнево-красный раствор аквокислоты (т. е. кислоты, образующейся за счет комплексного присоединения воды к нейтральной соли), дающей с ионами Аg• желтый осадок труднорастворимой серебряной соли — Аg2[ОАuСl3].
Наиболее обычным соединением трехвалентного золота является з о л о т о х л о р и с т о в о д о р о д н а я кислота, выделяющаяся в виде кристаллогидрата Н[АuСl4]·4Н2O при упаривании раствора золота в насыщенной хлором соляной кислоте. Из ее солей наиболее важен желтый х л о р а у р а т натрия — Nа[AuСl4]·2Н2О («золотая соль»). Как сама кислота, так и многие ее соли хорошо растворимы не только в воде, но и в некоторых органических растворителях (спирт, эфир). Очень малой растворимостью хлораурата цезия пользуются иногда для открытия этого элемента.
158-179
При сопоставлении элементов обеих подгрупп 1 группы между теми и другими можно наметить лишь немногие черты сходства. В частности, все металлы 1 группы отличаются высокой электропроводностью и образуют соединения, в которых они одновалентны. Однако Li и его аналоги т о л ь к о одновалентны, между тем как элементы подгруппы меди способны проявлять (а в случаях Сu и Аu даже предпочтительно проявляют) более высокую валентность. В этом отношении несколько ближе других элементов подгруппы меди к щелочным металлам стоит серебро.
180
Дополнения
15) Все элементы семейства меди кристаллизуются по типу куба с центрированными гранями (рис. Х11-38) при следующих ядерных расстояниях (пм): 256 (Сu), 289 (Аg), 288 (Аu). Электросопротивление всех трех металлов с повышением давления уменьшается (рис. Х111-40). Серебро лучше почти всех других металлов отражает лучи инфракрасной и видимой частей спектра:
Длина волны, нм...........1 000 500 100 50 40 31,6 30 20
Отражение лучей, %… 99 98,5 96 90 84 4 20 27
Интересна характерная для него прозрачность в ультрафиолетовой области около 31,60 пм. По отношению к инфракрасным лучам большой отражательной способностью обладает и золото.
16) Теплоты плавления рассматриваемых элементов равны 13,0 (Сu), 11,3 (Аg) и 12,6 (Аu) кДж/моль. Температура плавления меди последовательно повышается с ростом давления (до 20 тыс. ат). Объем серебра увеличивается при плавлении на 3,3 %.
17) При температурах кипения рассматриваемых элементов теплоты их испарения равны 305 (Сu), 255 (Аg) и 326 (Аu) кДж/моль. Помимо отдельных атомов, пары содержат и двухатомные молекулы (при температурах 1000-1300 °С и давлениях 10-3-10-6 мл рт. ст. отношение Э2: Э составляет около 1:1000), для которых имеются следующие данные:
Сu2 | Ag2 | Au2 | |||
| Ядерное расстояние, пм | 222 | 253 | |||
| Энергия диссоциации, кДж/моль | 192 | 167 | 222 |
Из сопоставления с аналогичными величинами для щелочных металлов вытекает, что ядерные расстояния в данном, случае меньше, а энергии диссоциации больше. Если отвлечься от золота (особое положение которого обусловлено лантанидным сжатием), то данные для меди, серебра [d(АgАg) » d(AuАu)] и щелочных металлов довольно хорошо укладываются на одну кривую зависимости энергий диссоциации от ядерных расстояний. Это говорит в пользу однотипности валентных (s) связей между атомами у элементов обоих подгрупп первой группы. В противоположность производным Аg и Аu, летучие соединения Сu окрашивают несветящееся пламя горелки (в сине-зеленый цвет).
18) Летучесть серебра при высоких температурах используется в ракетной технике. Как видно из показанной на рис. Х!11-41 схемы сопла, его внутренняя графитовая обкладка (А) в наиболее горячей зоне защищается от выгорания пластиной плотного вольфрама (Б) с внутренней полостью (В), которую заполняет пористый вольфрам, пропитанный серебром. Испарение последнего охлаждает вольфрам, что дает ему возможность противостоять действию стремительного потока газа, нагретого примерно до 3000 °С.
19) Основными потребителями м е д и являются электротехника и металлургия. В первой из них медь используется главным образом для изготовления электрических проводов, во второй — для выработки разнообразных сплавов с другими металлами. Идущая для Электротехнических целей медь должна быть достаточно чистой, так как большинство примесей снижает ее электропроводность (рис. Х1!1-42). Соединения меди применяются для борьбы с вредителями сельского хозяйства, в промышленности минеральных красок, стекольной и др. Мировая выработка этого металла составляла в 1800 г. 20 тыс. т, а в 1900 г. — 500 тыс. т. В настоящее время ежегодно добывается около 5 млн. т меди.
20) С е р е б р о ранее служило главным образом для выделки разменной монеты, домашней утвари и украшений. В настоящее время большой спрос на него предъявляют некоторые отрасли промышленности (электротехническая и др.). Его применяют также для изготовления частей заводской аппаратуры некоторых химических производств. В лабораториях серебряными тиглями пользуются для плавления щелочей, при высоких температурах действующих разъедающее почти на все другие материалы. Соединения Аg находят применение преимущественно в фотографической промышленности и медицине. Мировая выработка серебра составляла в 1800 г. 800 т, а в 1900 г. 5500 т. В настоящее время его ежегодно добывается около 10 тыс. т (без СССР). Приблизительно 2/3 всего серебра получено при комплексной переработке полиметаллических сернистых руд.
21) 3 о л о т о является основой денежной системы большинства стран, и поэтому громадные количества его хранятся без движения в подвалах банков для обеспечения выпущенных в обращение бумажных денег. Помимо этого, золото находит довольно широкое применение в электротехнической промышленности, употребляется для выделки различных предметов роскоши, золочения других металлов и т. и. Соединения золота используются главным образом в фотографии и в медицине. Мировая добыча золота составляла в 1800 г. 18 т, а в 1900 г. 400 т. В настоящее время его ежегодно добывается около 1500 т. Золотой запас всех зарубежных стран оценивается в 50 тыс. т.
22) Ниже приводится приблизительный состав некоторых наиболее известных сплавов на основе меди: б р о н з а (обычная) — 90% Сu, 10% Sn; т о м п а к — 90 % Cu, 10 % Zn; манганин — 86 % Сu, 12 % Мn, 2 % Ni; м е л ь х и о р — 68 % Cu, 30 % Ni, 1 % Мn, 1 % Fе; н е й з и л ь б е р — 65 % Сu, 20 % Zn, 15 % Ni; л а т у н ь (обычная) 60 % Сu, 40 % Zn; к о н с т а н т а н — 59 % Сu, 40 % Ni, 1 % Мn. Для выделки украшений пригоден золотистый сплав, состоящий из 85 % Сu, 13 % Zn и 2 % Sn. Еще более похож по виду на золото сплав состава 90 % Сu, 7,5 % Al и 2 % Au. Разменная монета СССР содержала 95 % Сu и 5 % Аl (до 5 коп.) или 80 % Сu и 20 % Ni (10 коп. и выше). Легко растирающийся в порошок сплав состава 50 % Cu, 45 % Al, 5 % Zn («сплав Деварда») иногда применяется в качестве восстановителя. Из воды сплав этот выделяет водород уже на холоду.
23) Международные золотой и серебряный монетные сплавы состоят соответственно из 90 % Аu или Аg и 10 % Сu. Следует отметить, что монеты такого состава в настоящее время почти не имеют хождения. Для выделки различных украшений и предметов роскоши обычно употребляются сплавы Аg с Сu, или Аu с Аg и Сu, состав которых (п р о б у) выражают числом весовых частей драгоценного металла в 1000 частях сплава. Например, для золотых зубных протезов применяются сплавы с пробой 900 или 916, а для ювелирных изделий — с пробами 958, 750, 583, 500, 375. Золотые сплавы с пробой 583 и выше не поддаются действию азотной кислоты. В СССР сохранилось еще много изделий, помеченных прежним русским способом — числом золотников драгоценного металла в фунте (96 золотников) сплава. В этом выражении золотые изделия изготовлялись обычно 5б, а серебряные — 84 пробы. Интересно отметить, что в древнем Египте серебро одно время (около 2500 лет до н. э.) ценилось дороже золота. Современная цена золота примерно в 40 раз выше, чем серебра.
25) Сплав Аu с 25 % Ag имеет зеленый, а с 20 % Ni — белый цвет. Сплавы золота с серебром, содержащие более 50 ат. % Аu, не поддаются действию НNО3. При меньшем содержании золота серебро растворяется и тем самым отделяется от золота. Это Было известно еще алхимикам. Так, в относящемся к Х111 веку (и приписываемом богу Гермесу) сочинении «Книга квинтэссенции» дается следующее указание: «Теперь научу вас способу разделения золота от серебра. Если хотите одно отвести от другого, то положите смесь их в крепкую водку, сделанную из купороса и селитры серебро растворится, но не золото». И далее: «А если вы хотите растворить золото, то положите его в крепкую водку, к ней прибавьте нашатыря: эта жидкость наверняка растворит золото». Под «крепкой водкой» здесь понимается азотная кислота (которая с нашатырем образует царскую водку).
26) Обычное поверхностное изменение меди на воздухе идет в основном по уравнению:
2 Cu + O2 + Н2О + СО2 = (СuОН)2СО3
Если воздух содержит примесь сернистого газа, то параллельно может протекать процесс по схеме:
8 Сu +5 O2 + 6 Н2О + SO2 = 2 [СuSO4·3Cu(OН)2]
В присутствии Н2S образуется черная пленка, состоящая из Сu2S и СuS.
27) Заметное взаимодействие меди с кислородом наступает около 200 °С. Идет оно по схеме Сu ® Сu2О ® СuО, причем монооксид меди начинает образовываться лишь после достижения слоем гемиоксида достаточной толщины (более 250 нм). Обусловлено это тем, что процесс последовательного окисления развивается в основном за счет диффузии (сквозь пленку оксида) атомов меди к кислороду, а не обратно. Таким образом, поверхностно окисленная до СuО медь содержит промежуточный слой Сu2О. Следует отметить, что первоначальная стадия окисления малозаметна, так как Сu2О почти не отчается по цвету от меди. Отдельные грани монокристалла Сu окисляются с несколько (до 20 %) различной скоростью.
28) Под нормальным давлением кислорода с е р е б р о не только с ним практически не реагирует, но и крайне мало его растворяет. Напротив, жидкое серебро растворяет кислород довольно хорошо. Поэтому при затвердевании Аg происходит выделение из него газообразного кислорода, иногда сопровождающееся разбрызгиванием металла.
Для з о л о т а имеются указания на обратное поведение. Так, отмечалось, что объемная растворимость кислорода в жидком Au имеет порядок лишь 0,2, тогда как твердое Аu способно при 450 °С поглотить до 48 объемов кислорода. Если это действительно так, то подобное различие между Аg и Аu очень интересно.
29) Оптическими исследованиями установлено, что на воздухе поверхность золота несет тончайший адсорбированный слой кислорода. У серебра подобным же образом обнаружено наличие оксидной пленки толщиной до 120 нм. При умеренном (300-400 °С) нагревании этого металла в атмосфере О2 образуется и более толстая поверхностная пленка оксида, а под избыточным давлением кислорода (20 ат) серебро может быть окислено даже нацело. Золото непосредственно не соединяется не только с кислородом, но также с серой и селеном. Однако жидкий теллур растворяет его с образованием теллуридов.
30) Взаимодействие элементов подгруппы меди с г а л о г е н а м и сильно ускоряется в присутствии влаги, при нагревании и под действием света. Продуктами взаимодействия меди и серебра (при избытке галогена) являются соответствующие галогениды СuII и АgI (кроме АgF2). Интересны особенности поведения золота. Соединение его с фтором идет лишь медленно даже при 300-400 °С. Для реакции с хлором наиболее благоприятен температурный интервал 150-300 °С, причем вблизи его нижней границы образуется преимущественно АuСl3, а вблизи верхней — АuCl. Бром является единственным галогеном, с которым золото взаимодействует уже при обычной температуре. В этих условиях образуется смесь АuВr3 с АuВr, а выше 60 °С — только АuВr. Для взаимодействия с иодом благоприятен интервал 50-110 °С, причем получается только АuI.
31) При взаимодействии меди с горячей концентрированной серной кислотой на основной процесс
Сu + 2 Н2SO4 = СuSO4 + SO2 + 2 Н2О
в большей или меньшей степени накладывается параллельная реакция:
5 Сu + 4 Н2SO4 + 3 СuSO4 + СuS + 4 Н2О
Относительное ее значение максимально около 100 °С, а при дальнейшем повышении температуры уменьшается и выше 270 °С сходит на нет.
32) При нагревании металлического серебра в атмосфере, хлористого водорода имеет место обратимая реакция:
2 Аg + 2 НСl = 2 АgСl + Н2 + 71 кДж
Равновесие быстро устанавливается уже при 200 °С. Если проводить процесс в замкнутом сосуде под атмосферным давлением, то при 600 °С газовая смесь содержит по объему 92,8 % НСl и 7,2 % Н2, а при 700 °С соответственно 95 % и 5 %. Аналогично протекает взаимодействие при тех же условиях в случае Сu.
35) На реакции по схеме АgСl + е-Ы АgЇ + Сl’ основана работа иногда применяемого в качестве электрода сравнения х л о р с е р е 6 р я н о г о э л е к т р о д а. Он состоит из опущенной в раствор КСl пластинки металлического серебра, покрытой слоем АgСl, и имеет нормальный потенциал +0,22 в.
36) При восстановлении соединений золота оно часто не выпадает в осадок, а образует коллоидные растворы (с отрицательным зарядом частиц). Золи эти интересны своей красивой окраской, меняющейся с увеличением размеров частиц по ряду цветов: бледно-розовый, красный, пурпурный, синий, фиолетовый, коричнево-черный. Образование пурпурного золя при действии SnСl2 на очень разбавленные (до 0,01 мг/л) растворы соединений золота используется иногда при анализах для открытия этого элемента. Золи золота были известны еще в ХVII веке.
37) Интересное применение находит получаемое восстановлением мелко раздробленное серебро в санитарной технике и медицине. Как показывает опыт, ион Аg• обладает исключительно сильно выраженными бактерицидными (убивающим бактерии} свойствами, Например, выдержанная некоторое время в серебряных сосудах вода может затем вне контакта с воздухом сохраняться без загнивания неограниченно долго, так как она оказывается достаточно стерилизованной уже той ничтожной концентрации иона Аg, которая в ней создается при соприкосновении с металлическим серебром. Это было известно еще в древности. Так, около 2500 лет тому назад персидский царь Кир пользовался серебряными сосудами для хранения питьевой воды во время своих военных походов.
38) Накопление ионов Аg• протекает тем быстрее, чем больше поверхность соприкосновения воды с металлом. Для максимального увеличения этой поверхности с наименьшей затратой металла целесообразно осаждать последний очень тонким слоем на зернах обычного песка и затем фильтровать воду сквозь слой такого посеребренного песка. Подобным образом могут быть созданы удобные походные фильтры для обеззараживания воды. С другой стороны, перевязки с применением полученной тем же путем «серебряной марли» или «серебряной ваты» (либо просто присыпки порошком коллоидного серебра) хорошо действуют при лечении некоторых кожных заболеваний, трудно заживающих язв и т. д. Покрытие поверхностных ран серебряными пластинками практиковалось уже в древнем Египте.
39) «Серебряная вода» может служить для обеззараживании и консервирования некоторых пищевых продуктов. Следует отметить, что более быстрым и удобным способом ее получения является контакт воды не с металлическим Аg, а с хлористым серебром. Создающаяся в насыщенном растворе этой соли концентрация Аg• составляет около 1 мг/л. тогда как нижний предел бактерицидного действия серебра оценивается концентрацией порядка 10-6 мг/л. Следовательно, насыщенный раствор АgСl без потери его бактерицидности может быть разбавлен в 100 и более раз.
40) Очистку больших количеств воды на основе использования бактерицидного действия иона Ag• особенно удобно проводить электрохимическим путем. Для этого достаточно иметь небольшой источник постоянного тока и две серебряные пластинки в качестве электродов. Током силой в 10 мА (при напряжении около 1,5 в) можно осуществить стерилизацию 4000 л воды за час.
41) Несколько неясен вопрос о возможной токсичности «серебряной воды». Прямых указаний на это нет, но по санитарным нормам США содержание Аg в питьевой воде не должно превышать 0,05 мг/л. Очевидно, что при этом подразумевается постоянное пользование ею.
42) При длительном поступлении в организм избыточных доз серебра развивается аргирия, внешне выражающаяся серой окраской слизистых оболочек и кожи (преимущественно на освещенных участках тела), обусловленной отложением частичек восстановленного серебра. Какие-либо расстройства самочувствия заболевших аргирией наблюдаются далеко не всегда. Вместе с тем отмечалось, что они не подвержены инфекционным заболеваниям.
43) Гемиоксид меди встречается в природе и может быть получен прокаливанием Сu при ограниченном доступе воздуха. Теплота его образования из элементов, равна 167 кДж/моль. В кристаллах Сu2О каждый атом кислорода тетраэдрически окружен четырьмя атомами меди, а каждый из последних имеет два соседних атома кислорода (рис. Х111-16). По отношению к нагреванию гемиоксид меди весьма устойчив и плавится при 1236 °С без разложения. Напротив, Аg2О (теплота образования 29 кДж/моль) и Au2О начинают распадаться на элементы уже около 100 °С. Давление диссоциации Аg2О достигает 1 атм при 182 °С.
44) При наличии небольшого избытка кислорода (против формулы Сu2О) гемиоксид меди является полупроводником р-типа. На его кристаллах были наиболее детально учены свойства возникающих в полупроводниках и диэлектриках (например, под действием света) элементарных электронейтральных возбуждений — э к с и т о н о в. Последние модельно трактуются как способные перемещаться по объему кристалла более или менее тесные сочетания электрона и «дырки».
45) Интересным применением Сu2О является использовании ее в паре с металлической медью для изготовления «купроксных» выпрямителей переменного тока. Работа основана на том, что поток электронов проходит только от Сu к Сu2О, но не обратно.
46) При восстановлении производных двухвалентной меди в щелочной среде выпадает желтый осадок, по-видимому, представляющий собой в основном не СuОН, а коллоидную Сu2О. По мере довольно быстро протекающего укрупнения ее частиц цвет меняется на красный. Для произведения растворимости СuОН дается значение 2·10-15.
47) В индивидуальном состоянии белый АgОН может быть, по-видимому, получен взаимодействием спиртовых растворов АgNО3 и КОН при -50 °С. Растворимость АgОН в воде при обычных условиях составляет приблизительно 2·10-4 моль/л. Осаждение его из растворов идет около рН = 9. Диссоциации АgОН по основному и кислотному типам отвечают, соответственно, следующие константы: К = 5·10-3 и К = 8·10-13. Таким образом, на каждую молекулу АgОН, диссоциированную по кислотному типу, приходятся миллиарды диссоциированных по типу основания.
48) Растворимость АgОН в крепких растворах сильных щелочей значительно выше, чем в воде. Растворы содержат а р г е н т и т ы типа М[Ag(OH)2] которые в индивидуальном состоянии не выделены. Сухим путем были получены аргентиты составов Nа3АgО2, Nа4Ag2О3 и Nа2Аg4О3, однако весьма вероятно, что в действительности они представляют собой сплавы оксидов.
Напротив, тоже синтезированные сухим путем продукты состава МОЭ (где М — щелочной металл, а Э — элемент подгруппы меди) являются индивидуальным соединениями. Кристаллы этих бесцветных (КОСu, КОАg) или желтых (СsОАg, СsОАu) веществ слагаются из тетрамеров М4[Э4O4].
49) Темно-фиолетовый АuОН может быть получен слабым подогреванием взвеси АuВr в растворе КОН. При избытке щелочи АuОН образует синий золь, но вместе с тем и частично растворяется. Последнее обусловлено, вероятно, образованием
а у р и т о в М[Аu(ОН)2], которые в твердом состоянии не выделены. Осторожным нагреванием (не выше 200 °С) моногидрат золота может быть нацело переведено в Аu2О.
50) Во влажном состоянии соли Сu+ постепенно окисляются кислородом воздуха, а большинство соединений Аu+ довольно быстро распадается на соответствующие производные трехвалентного золота и металлическое Аu (по схеме 3 Аu+ = Аu3+ + 2 Аu). Подобный же распад (по схеме 2 Сu+ = Сu2++ Сu) имеет место и у некоторых производных одновалентной меди.
54) Ввиду малой растворимости хлористых, бромистых и иодистых солей серебра их творожистые осадки образуются во всех тех случаях, когда раствор одновременно содержит ионы Аg• и галогена. Сложнее обстоит дело в случае меди, не дающей легкорастворимых солей в одновалентном состоянии. Здесь приходится исходить из производных двухвалентной меди, восстанавливая их нагреванием с металлической Сu (например, по схеме: CuCl2 + Cu = 2 СuСl).
55) Галлоидные соли одновалентного золота удобно получать разложением при умеренном нагревании соответствующих производных трехвалентного Аu (по схеме АuГ3 = Au + Г2) или их осторожным восстановлением. В присутствии воды эти почти нерастворимые галогениды подвергаются дисмутации по схеме 3 АuГ = АuГ3 + 2 Аu. На холоду процесс протекает медленно, но значительно ускоряется при нагревании (и под действием света). По ряду: AuCl-AuBr-AuI тенденция к дисмутации уменьшается и зеленовато-желтый иодид (который может быть при слабом нагревании получен также по схеме 2 Аu + I2 = 2 АuI) выше 100 °С начинает распадаться непосредственно на элементы.
60) Весьма практически важным свойством труднорастворимых галогенидов Аg является постепенно идущий под действием света распад их на металлическое серебро и свободный галоген по схеме 2 АgГ = 2 Аg + Г2, Распад этот вызывается главным образом лучами сине-фиолетовой части спектра, тогда как красные лучи оказываются практически неактивными. Светочувствительность галогенидов серебра изменяется по ряду
АgВr>АgСl>АgI. При охлаждении она сильно уменьшается. Так, при температуре жидкого воздуха чистое АgСl заметно не разлагается даже в результате длительного освещения. Кристаллы AgСl хорошо пропускают инфракрасное излучение (особенно более 80 % — в интервале от 5 до 20 мк).
61) Протекающие под действием света химические процессы носят название фотохимических реакций. В зависимости от того, является ли сам рассматриваемый процесс экзо- или эндотермическим, значение световых лучей для его протекания существенно различно. В первом случае (например, при образовании НСl из элементов — свет нужен только для первоначального возникновения процесса, а дальше он самопроизвольно идет и в темноте. Напротив, во втором случае (примером которого может служить распад галогенидов Аg) процесс протекает только в меру затраты на него энергии световых лучей и при устранении освещения прекращается.
62) Фотохимическая чувствительность галогенидов серебра была использована, в частности, для создания светочувствительного стекла. Заключенные в стеклянной массе мельчайшие частицы АgГ под действием сильного света разлагаются с образованием металлического серебра (и свободного галогена), отчего стекло темнеет. Так как серебро и галоген пространственно не отделены друг от друга, при ослаблении света наступает обратная реакция и стекло вновь становится прозрачным. Подобные светочувствительные стекла, по-видимому, перспективны для использования в электронно-вычислительной технике.
63) На светочувствительности галогенидных солей серебра основано использование для фотографии. Фотографические пленки состоят в основном из светочувствительного слоя тонкой взвеси галогенида Аg (чаще всего — АgВr) в желатине, нанесенного на целлулоид. Обычная толщина такого слоя составляет около 20 мк, а диаметр зерен АgГ от 0,1 до 2 мк.
Освещение пленки ведет к распаду содержащегося в ней галогенида Аg, причем галогенид химически связывается желатиной, а серебро образует мельчайшие зародышевые кристаллики. Последних на данном участке поверхности возникает тем больше, чем более сильному освещению этот участок подвергался. Таким образом, несмотря на внешнюю однородность находившейся под кратковременным действием света пленки, на ней уже содержится «скрытое изображение» фотографируемого предмета.
Чтобы сделать его видимым, пленку подвергают операции проявления, заключающейся в дальнейшем восстановлении галогенида серебра до свободного Аg химическим путем. В качестве проявителя пользуются обычно каким-либо органическим восстановителем. Существенно важно для процесса проявления то обстоятельство, что восстановление галогенида серебра быстрее всего протекает в непосредственном соседстве с уже имеющимися зародышевыми кристалликами металлического Аg. Обусловлено это, по-видимому, адсорбцией на них проявителя, с одной стороны, и ролью зародышей в качестве центров кристаллизации для вновь выделяющегося металлического серебра — с другой.
Достигнув при помощи проявления достаточной четкости видимого изображения, пленку фиксируют для того, чтобы уничтожить ее чувствительность к дальнейшему действию света. Фиксаж осуществляется путем извлечения из светочувствительного слоя оставшихся неразложенными галогенидов серебра. В качестве фиксирующего агента обычно пользуются раствором гипосульфита, легко растворяющим галогениды Аg за счет комплексообразования по схеме:
2 Nа2S2О3 + АgГ = Nа3[Аg(S2O3)2] + NаГ
(константа нестойкости комплексного иона [Аg(S2О3)2]’” равна 4·10-14).
Получаемое путем проявления и фиксирования устойчивое на свету видимое изображение — н е г а т и в — является обратным истинному, так как темные места на нем отвечают светлым местам фотографируемого объекта и обратно. Для получения истинного изображения негатив накладывают на другую пленку или бумагу со светочувствительным слоем и подвергают действию света. Так как свет легче проходит сквозь более светлые места негатива и труднее — сквозь более темные, отношение между светом и тенью меняется при таком «печатании» снимков на обратное и становится отвечающим сфотографированному объекту. Отпечатанный снимок — п о з и т и в — подвергают затем проявлению и фиксированию (или только последнему, если светочувствительный слой был рассчитан на получение видимого изображения непосредственно фотохимическим путем).
64) Недавно было предложено создавать светочувствительный слой путем осаждения на стекле паров АgВr в вакууме. Толщина напыленного таким образом слоя не превышает 4 мк, а зернистость его практически не сказывается при любых увеличениях. Время последующей обработки негатива (проявления и т. д.) исчисляется всего лишь секундами.
65) Путем введения в светочувствительный слой специальных добавок удается не только резко повышать его общую чувствительность (вплоть до времени экспозиции 10-12 сек) или избирательную восприимчивость к отдельным лучам спектра (в том числе инфракрасным), но и создавать слои, приобретающие под действием света и последующего проявления определенную окраску. На этом основана ц в е т н а я ф о т о г р а ф и я, которая обычно использует возможность получения любого цвета путем комбинирования в соответствующих пропорциях трех элементарных цветов — красного, зеленого и синего.
66) Интересно, что подобным же образом строятся и наши вкусовые ощущения. Язык содержит рецепторы четырех основных типов, отвечающих сладкому, соленому, кислому и горькому вкусам. Различные комбинации этих элементарных вкусов и создают всю гамму вкусовых восприятии человека. Интересно, что вкусовая чувствительность людей с возрастом не понижается, а повышается.
67) При нагревании все рассматриваемые галогениды меняют свою окраску на более темную. Например, СuСl при 178 °С синеет, а хлористое серебро становится при плавлении оранжево-желтым. Так как расплавленное АgСl хорошо пристает к стеклу, кварцу и металлам, им удобно пользоваться для создания газонепроницаемых соединений между отдельными частями аппаратуры (следует лишь иметь в виду, что при переходе из жидкого в твердое состояние объем АgСl увеличивается. Более легкоплавки пригодные для той же цели сплавы состава 40 вес. '%, АgСl и 60 — ТlСl (т. пл. 210 °С) или 31 вес. % АgСl, 46 — АgI и 23 — ТlСl (т. пл. 131 °С). Расплавленный АgСl несколько растворяет металлическое серебро (0,03 мол. % при 490 °С и 0,06 мол. % при 700 °С).
68) Выше 1000 °С все галогениды Сu+ и Аg+ заметно летучи, причем при дальнейшем нагревании одни кипят без разложения (СuСl — при 1359, СuВr — при 1345, СuI — при 1336, АgСl — при 1545 °С), другие начинают распадаться на элементы еще по достижения точки кипения (например, АgI). Еще легче (до достижения точки плавления) распадаются при нагревании галогенидные соли одновалентного золота. По ряду Сl-Вr-I термическая устойчивость рассматриваемых соединений заметно уменьшается.
71) В сухом состоянии галогениды Сu+, Аg+, Au+ легко присоединяют газообразный NН3 с образованием комплексных соединений, из которых наиболее богатые аммиаком отвечают составу [Э(NН3)3]Г. Аммиакаты с еще большим содержанием NН3 могут быть получены при пользовании жидким аммиаком. Например, АuСl в этих условиях образует комплекс состава АuСl·12NH3.
72) Водный аммиак растворяет большинство галоидных солей Сu+ и Ag+ с образованием бесцветных комплексных соединений, содержащих преимущественно ионы [Э(NН3)2]+. Интересно, что по ряду АgСl-АgВr-АgI растворимость галогенидов в водном аммиаке очень быстро возрастает (100 г насыщенного при 0 °С раствора содержат 0,28 г АgСl, 2,35 г АgВr или 84,1 г AgI). Для АuСl известен бесцветный аммиакат состава АuСl·NН3, образующийся при взаимодействии АuСl с NН4ОН (и последующем упаривании раствора). Он нерастворим в воде, но растворяется в избытке NН4OН (вероятно, вследствие образования [Аu(NH3)2]Cl. При нагревании выше 150 °С этот аммиакат разлагается. Иодистое золото образует с жидким аммиаком комплекс состава [Аu(NН3)6]I.
73) Подобно солям Аg+, легко растворяется в аммиаке и Аg2O. В результате растворения образуется комплексное основание [Аg(NН3)2]ОН, диссоциирующее приблизительно в той же степени, как NаОН. Константа нестойкости иона [Ag(NH3)2]• равна 6·10-8. Аналогичный комплекс одновалентной меди сам по себе еще устойчивее (К = 1·10-11), но легко окисляется кислородом воздуха. Следует отметить, что раствор аммиачного комплекса серебра нельзя хранить (так как при стоянии из него выделяется весьма взрывчатый осадок).
74) При помощи восстановления аммиачных растворов солей серебра могут быть получены плотно пристающие к стеклу тонкие пленки металлического Аg. На этом основании производство з е р к а л. Осажденная на стекле блестящая серебряная пленка для защиты от внешних воздействий покрывается сверху лаком. Обычно для серебрения стекла применяются два свежеприготовленных раствора, примером получения которых дается ниже. (А) К раствору 6 г АgNО3 в 100 мл воды добавляют водный аммиак до растворения первоначально образующегося осадка, затем 70 мл 3 %-ного раствора NаОН и снова водный аммиак до полного прояснения раствора (без избытка). Последний разбавляют водой до 500 мл. (Б) Раствор 1,3 г глюкозы в 25 мл воды (к которой добавлена одна капля конц. HNО3) кипятят 2 мин, охлаждают и разбавляют равным объемом спирта. Перед самым употреблением растворы А и Б смешивают в соотношении 10:1. Плотная пленка серебра осаждается на стекле (предварительно тщательно очищенном обработкой горячей смесью НNО3 + К2Сr2О7, затем дистиллированной водой и, наконец, спиртом) примерно через 1/2 ч. При необходимости получения более толстого слоя Аg обработку повторяют со свежими порциями растворов еще один или два раза. Образовавшийся осадок серебра промывают водой и спиртом.
75) Раствором AgNО3 в водном аммиаке иногда пользуются для нанесения несмываемых при стирке меток на белье. Проявление и закрепление метки достигается путем немедленного проглаживания помеченного места горячим утюгом. В случае необходимости снятия такой метки это может быть осуществлено обработкой ее крепким раствором KI с последующей промывкой водой.
78) Малорастворимые галогениды серебра растворимы в крепких горячих растворах АgNО3 (а также AgСlO4, и АgF). Из подобных растворов могут быть выделены двойные соли АgNО3 и соответствующего галогенида Аg, характеризующиеся определенными температурами плавления (°С):
АgNО3·АgСl АgNО3·АgВr АgNО3·АgI 2АgNО3·АgI
160 182 94 105
В рассматриваемых двойных соединениях комплексообразователем является ион галогена: строение их отвечает формулам [ГАg2]NО3 и [ГАg3](NО3)2. Для констант нестойкости ионов [ГАg2]• и [ГАg2]•• даются следующие значения: 3·10-5 и 1·10-5 (Сl), 9·10-8 и 7·10-9 (Вr), 1·10-11 и 2·10-14 (I).
79) Бесцветные аммиачные и солянокислые растворы СuСl на воздухе быстро окрашиваются (соответственно — в синий или зеленый цвет) вследствие постепенно идущего окисления Сu+ до Сu2+. В газовом анализе этими растворами часто пользуются для улавливания монооксида углерода. Последний поглощается ими на холоду и вновь выделяется при нагревании. Из насыщенных оксидом углерода растворов может быть выделено бесцветное соединение состава СuСl·СО·2Н2О. Соответствующий безводный продукт присоединения (СuСl·СО) образуется только под большим давлением. Он представляет собой рыхлый белый порошок, давление СО над которым при 0 °С составляет 67 мм рт. ст. Аналогичные производные СuВr и СuI еще менее устойчивы. Подобное же по составу бесцветное соединение золота (АuСl·CO) образуется в результате взаимодействия АuСl3 и СО при 90 °С. Для серебра карбонильные производные не получены.
80) К галоидным солям Сu+, Аg+ и Аu+ близко примыкают их ц и а н и д ы: белые СuСN и АgСN и желтый АuСN. Температура плавления СuСN лежит при 473 °С (в атмосфере азота), а цианиды серебра и золота разлагаются еще до ее достижения. Под давлением около 73 тыс. атм температура плавления АgСN равна 443 °С (а ее экстраполяция к обычному давлению дает 346 °С). В отличие, от галогенидных солей серебра АgСN не темнеет под действием света. Кристалл его слагается из бесконечных линейных цепей типа •••Аg•••СєN•••Аg•••СєN•••, т. е. серебро оказывается связанным и с углеродом, и с азотом цианидных анионов. Аналогичная структура установлена и для цианида одновалентного золота. Интересно, что расстояние между соседними атомами Аg в таких цепях (509 пм) значительно меньше, чем между соседними атомами Аg (526 пм). Частота валентных колебаний связи CєN в АgСN (2178 см-1) значительно выше, чем в ионной структуре NаСN (2080 см-1).
81) Все три соли почти нерастворимы в воде и разбавленных кислотах, но более или менее хорошо растворяются в веществах, образующих с ионами рассматриваемых элементов комплексные соединения (NН3, Nа2S2О3 и др.). Особенно легко идет их растворение в присутствии цианидов щелочных металлов. При этом получаются весьма устойчивые, легкорастворимые в воде бесцветные комплексные соли типа М[Э(СN)2].
83) Нормальные потенциалы (В) для протекающих в щелочной среде процессов
Э+2 СN’Ы [Э(СN)2]’+ е-
равны -0,43 (Сu), -0,31 (Ag) и -0,61 (Аu), т. е. близки к потенциалу водородного электрода. Несмотря на принципиальную обратимость при этих условиях реакций по схеме Э + Н•Ы Э•+ Н, вытеснение водорода заметно наблюдается лишь в случае меди (склонной к образованию ионов [Cu(СN)3]” и [Сu(СN)4]’”). Однако в присутствии окислителей происходит довольно быстрое растворение также серебра и золота, так как равновесие Э + Н•Ы Э· + H смещается вправо за счет связывания уже не только Э•, но и водорода (путем окисления его до воды). При доступе кислорода воздуха золото и серебро растворяются в NаСN по суммарному уравнению:
4 Э + 8 NаСN +2 Н2О + O2 = 4 Nа[Э(СN)2] + 4 NаОН
В качестве промежуточного продукта при этом, по-видимому, образуется пероксид водорода, расходуемый затем на дальнейшее окисление. Растворами комплексных цианидов Аg и
Аu часто пользуются для электролитического серебрения и золочения других металлов (главным образом меди).
84) Для р о д а н и д н ы х производных Сu+ и ее аналогов характерна связь катиона с анионом через серы. Однако кристаллы АgSСN слагаются из бесконечных зигзагообразных цепей типа… АgSСN… АgSСN… (с углом 104° при атоме серы), в которых Ag+ имеет контакты не только с серой [d(АgS) = 243 пм], но и с азотом соседней молекулы [d(AgN) = 222 пм]. Простые роданиды Сu+ и Аg+ представляют собой белые вещества, почти не растворимые в воде, но при избытке ионов SСN’ растворяющиеся с образованием комплексных соединений. Из них для серебра известны типы М[Аg(SCN)2], M2[Ag(SCN)3] и М3[Аg(SСN)4], а также (NН4)5[Аg(SСN)6]. Для меди получены Сs[Сu(SCN)2] и Nа3[Сu(SСN)4]·4Н2О. Последняя соль интересна тем, что в ее концентрированном растворе фильтровальная бумага сильно набухает, а при последующем нагревании образует прозрачный раствор, который по охлаждении застывает в гель. Для константы нестойкости ионов [Э(SСN)2]’ даются значения 1·10-11 (Сu), 3·10-8 (Аg) и 1·10-25 (Au). Роданистые производные Аu+ малоустойчивы и постепенно разлагаются с выделением металлического золота.
85) Из других солей Сu+, Аg+ и Аu+ практически приходится иметь дело почти исключительно с производными серебра. Данные по растворимости (моль/л Н2О) некоторых солей Ag при обычных условиях сопоставлены ниже:
ClO4- | NO3- | ClO3- | CH3COO- | ClO2- | SO42- | NO2- | BrO3- | SeO42- |
| 27 | 15 | 5·10-1 | 7·10-2 | 3·10-3 | 3·10-2 | 3·10-2 | 9·10-3 | 1·10-3 |
CO32- | CrO42- | N3- | C2O42- | IO3- | PO43- | CN- | SCN- | S2- |
1·10-4 | 7·10-5 | 5·10-5 | 1·10-4 | 2·10-5 | 2·10-5 | 2·10-6 | 1·10-6 | 6·10-18 |
Как видно из приведенного сопоставления, большинство солей серебра малорастворимо в воде.
86) В крепких растворах соответствующих солей щелочных металлов труднорастворимые соли Аg обычно более или менее легко растворяются вследствие образования комплексных соединений. Последние характеризуются различной устойчивостью (например, константы нестойкости ионов [Аg(NО2)2]’ и [Аg(SO3)2]’” равны соответственно 6·10-3 и 2·10-9). Интересными комплексными производными серебра являются соли состава Сs3Ba[Ag(NО2)6]·2Н2О и (NН4)9[Аg(S2O3)4Х2] (где Х — Сl, Вr или SСN-), в которых Аg имеет, по-видимому, координационное число шесть.
87) Нитрат серебра (т. пл. 209,6 °С) широко используется при изготовлении зеркал и в медицине (часто в виде сплава 1 вес. ч. АgNО3 с 2 вес. ч. КNО3 под названием “ляпис”). Расплавленное АgNО3 может служить средой для криоскопии (криоскопическая константа 26,5 град/моль). Термическое разложение этой соли наступает лишь выше 350 °С. Для системы AgNО3-КNО3 установлено наличие плавящейся при 125 °С эвтектики (55,6 мол. % АgNО3). Азотнокислое серебро растворимо и в некоторых органических жидкостях. Примерами могут служить приводимые ниже данные (в молях AgNО3 на 100 молей растворителя при обычных условиях):
Н2О СН3ОН С2Н5OН СН3СОСН3 СН3СООСН3 С6Н5NН2
27 0,68 0,57 0,21 1,4 9,8
С5Н5N СН3CN
21,3 41,6
Как видно из этих данных, особенно хорошими растворителями АgNО3 являются азотсодержащие органические соединения. Для Сu+ и Аg+ нитраты не получены (однако некоторые производящиеся от СuNО3 комплексы известны).
88) Пропускание этилена при 0 °С в насыщенный водный раствор нитрата серебра сопровождается осаждением белого комплекса С2Н4·AgNО3. В индивидуальном состоянии он устойчив лишь ниже -30 °С (или под повышенным давлением этилена).
89) Энергия кристаллической решетки перхлората серебра оценивается в 690 кДж/моль. При нагревании эта бесцветная соль не плавится, а около 500 °С разлагается. Она легко растворима не только в воде, но и во многих органических жидкостях. Твердая фаза обычно содержит сольваты с растворителем типа АgClО4·nX (где X — молекула растворителя). Сольват АgСlO4·С6Н5СН3 устойчив лишь ниже 22,6 °С. Обращает на себя внимание почти одинаковая растворимость АgСlO4 в анилине и бензоле, а также исключительно высокая (для неорганического соединения) его растворимость в толуоле. Отмечалось, что при добавлении небольшого количества воды она еще увеличивается. Некоторые числовые данные для 25 °С приводятся ниже (растворимость в молях АgСlO4 на 100 молей растворителя):
| Растворитель | H2O | C5H5N | C6H5NH2 | C6H6 | C6H5CH3 |
| Растворимость | 48 | 10,0 | 2,4 | 2,0 | 42 |
| n | 1 | 4 | 6 | 1 |
Перхлорат серебра хорошо растворим также в глицерине, уксусной кислоте, нитробензоле и хлорбензоле. Раствор его в нитрометане обладает хорошей электропроводностью. В разбавленном (0,0002 М) бензольном растворе нитрат серебра мономерен и обнаруживает дипольный момент 10,7, но по мере повышения концентрации этот момент уменьшается (до 4,7 для 0,005 М раствора), что обусловлено, вероятно, частичной димеризацией АgClO4. Следует отметить, что сочетание этой соли с большинством органических молекул способны взрываться (при нагревании или ударе).
90) По рентгеноструктурным данным монокристалл АgClO4·C6H6 слагается из цепей типа •••Аg•••С6Н6••• Аg•••С6Н6••• и располагающихся в промежутках между ними ионов СlO4-. Расстояния от иона Аg+ до ближайшей связи С-С двух колец неодинаковы (250 и 263 пм). Интересно искажение бензольного кольца: d(СС) в ближайшей к Аg+ связи равно 135 пм, тогда как в остальных — 143 пм; из РССС два равны 116°, а остальные — 122°.
Еще большее искажение бензольного кольца наблюдается у комплекса С6Н6·СuАlСl4. Атом меди в его кристалле координирован тремя атомами хлора и одной связью СС (от атомов которой он находится на расстояниях 215 и 230 пм). Связь эта имеет длину 127 пм, а длины остальных (141-125-137-129-140) чередуются.
92) Желтоватый нитрит серебра может, по-видимому, существовать в двух формах: Аg-ОNО и Аg-NО2 (причем обычно получается их смесь). Так, при его взаимодействии с С2Н5I образуются примерно одинаковые количества этилнитрита и нитроэтана (тогда как КNО3 дает только этилнитрит). Термическое разложение нитрита серебра наступает уже выше 128 °С. В водном растворе соль эта умеренно диссоциирована (К = 2·10-2). На воздухе она медленно окисляется до нитрата.
93) Бесцветный АgСlO3 (т. пл. 230 °С) может быть получен, в частности, взаимодействием мелкораздробленного серебра с хлорноватой кислотой по схеме
6 Аg + 6 НСlO3 = АgСl + 5 АgСlO3 + 3 Н2О
Кислотами (даже уксусной) AgСlО3 разлагается с образованием АgСl и выделением кислорода, тогда как АgBrО3 по отношению к ним гораздо устойчивее, а АgIO3 не разрушается даже горячей серной или азотной кислотой. По всей вероятности, серебро в этих солях связано непосредственно с галогеном.
94) Белый сульфат серебра (т. пл. 660 °С) устойчив на воздухе и начинает разлагаться по схеме
Аg2SO4 = 2 Аg + SO2 + О2
лишь около 1000 °С. Растворимость его в серной кислоте значительно выше, чем в воде (вследствие образования более растворимого бисульфата — АgHSO4). Напротив, в растворе К2SО4 она ниже, чем в воде, что указывает на малую тенденцию иона Аg+ к комплексообразованию с ионами SO42-. Для константы диссоциации иона АgSО4’ дается значение 0,59.
95) Желтоватый карбонат серебра при нагревании выше 100 °С начинает разлагаться по схеме
Ag2CO3 + 84 кДж = Аg2О + СО2
(энергия активации распада составляет 96 кДж/моль, и давление СО2 в 1 атм достигается при 218 °С). Интересно строение гораздо лучше растворимой смешанной соли КАgСО3. кристаллы ее слагаются из ионов К+ и цепей, образованных ионами АgСО3-.
97) Ввиду довольно высокой стоимости серебра соединения его после использования обычно собирают в специальных банках. Для регенерации Аg из таких остатков их кипятят с гранулированным цинком в присутствии НСl, что сопровождается осаждением порошкообразного серебра. Последний отделяют от избытка Zn, обрабатывают при нагревании разбавленной НСl и затем промывают водой. При необходимости более тщательной очистки полученное серебро, после растворения в НNО3, выделяют в виде АgСl. Хлорид серебра смешивают с разбавленным NаОН и добавляют формалин. При нагревании на водяной бане Аg быстро выделяется в виде рыхлого черного порошка, который отделяют от жидкости и последовательно промывают разбавленной H2SO4, водой, раствором NН4OН и снова водой. Хлорид серебра может быть разложен также сплавлением с содой — реакция идет по уравнению:
4 АgСl + 2 Na2CО3 = 4 NаСl + 2 СО2 + O2 + 4 Аg
98) Из солей Сu+ ближе всего к рассмотренным ранее галогенидам, цианиду и роданиду стоит азид — СuN3. Бледно-зеленые кристаллы этой соли почти нерастворимы в воде (растворимость 5·10-9 моль/л), а под действием света она разлагается на элементы.
99) Сульфат одновалентной меди может быть получен при 200 °С по реакции
2 Сu + 2 Н2SO4 = Сu2SО4 + SO2 + 2 Н2О
Он представляет собой бесцветные кристаллы, устойчивые в сухом воздухе, но водой разлагаемые на СuSO4 и Сu. Соль эта является сильным восстановителем (например, гидроксиламин количественно переводится ею в аммиак).
100) Сульфит одновалентной меди может быть получен действием тока SO2 на кипящий концентрированный раствор Сu(СН3СОО)2 в уксусной кислоте. Реакция идет по уравнению
3 Cu(СН3СОО)2 + 2 SO2 + 3 Н2О = СuSO4 + Сu2SO3 + 6 СН3СООН
и сульфит осаждается в виде белого кристаллогидрата Сu2SO3·Н2О. Имеется указание на возможность получения и красной октамерной формы этого вещества — (Сu2SO3·Н2О)8. При нагревании на воздухе димедь-сульфит переходит в Сu2О. Он малорастворим воде, но с растворами сульфитов щелочных металлов образует растворимые комплексные сульфиты (устойчивые лишь при рН = 7ё8). Для констант нестойкости ионов СuSO3’, Сu(SO3)2’” и Сu(SО3)3””’ даются значения 1·10-8 3·10-9 и 4·10-10. Интересным производным иона СuSO3-, является красная соль Сu(СuSO3)2·2Н2О, осаждающаяся при пропускании тока SO2 в 10 %-ный раствор СuSO4.
104) Из производных Аu+, помимо рассмотренных выше, известии лишь немногие. Ближе всего к его галогенидам, цианиду и роданиду стоит а з и д — АuN3 (теплота образования из элементов — 280 кДж/моль). Эта бесцветная соль может быть получена по схеме:
АuСl3 + 3 NаN3 = 3 NаСl + АuN3 +3 N2
Она растворима в воде и очень взрывчата (даже в растворе). Нейтральный тиосульфат одновалентного золота неизвестен, но по схеме
АuI + 2 Nа2S2О3 = NаI + Nа3[Аu(S2О3)2]
может быть получен тиосульфатный комплекс, выделяющийся из раствора в форме бесцветного кристаллогидрата (с 2Н2О). Обработкой порошка золота в ацетонитрильной среде перхлоратом серебра был получен кристаллический комплекс состава [Аu(NССН3)2]СlO4.
105) Сульфиды Сu+, Аg+ и Аu+ в воде и разбавленных кислотах практически нерастворимы. Темно-серый Сu2S (т. пл. 1130 °С) встречается в природе и является важной промышленной рудой меди. Он может быть синтезирован путем действия только давления (порядка тысяч атмосфер) на смесь тонких порошков Сu и S. Интересным свойством Сu2S является резкое уменьшение электрического сопротивления с повышением давления: при 12 тыс. атм оно составляет всего 0,1 % от величины в обычных условиях. Взаимодействие Сu2S с раствором АgNО3 протекает по уравнению:
Сu2S + 4 АgNО3 = АgS + 2 Аg + 2 Сu(NО3)2
106) Черный Аg2S (т. пл. 825 °С) представляет собой особенно труднорастворимую соль и поэтому осаждается ионом S” из растворов всех соединений серебра. Образование Аg2S происходит также при действии Н2S (в присутствии влаги и кислорода воздуха) на металлическое серебро. Реакция по уравнению:
4 Аg + 2 Н2S + O2 = 2 Аg2SЇ + 2 Н2О
медленно идет уже в обычных условиях и вызывает постепенное потемнение серебряных предметов домашнего обихода.
В принципе взаимодействие между Аg и Н2S может происходить и с вытеснением водорода, так как из-за очень малой растворимости Аg2S потенциал серебра снижается до -0,71 В. Однако в отсутствие кислорода образование Аg2S протекало бы несравненно медленнее. При контакте с алюминием в разбавленном растворе соды сульфид серебра легко восстанавливается до металла.
107) Коричнево-черный (во влажном состоянии — серый) сульфид одновалентного золота при нагревании до 240 °С разлагается на элементы. Он может быть получен действием Н2S на подкисленный раствор К[Аu(СN)2]. Так как протекающая по схеме
2 [Аu(СN)2]’+ S” = Аu2S + 4 СN’
реакция его образования обратима, необходимо сильное насыщение раствора сероводородом. На обратимости аналогичной реакции также для Аg2S основан иногда применяемый метод извлечения серебра из его сернистых руд. Чтобы сместить ее равновесие влево, ионы S” удаляют в этом случае путем окисления их (кислородом продуваемого сквозь раствор воздуха) до ионов S2O3”.
108) Сульфиды одновалентных Сu, Аg и Аu частично растворимы в растворах сульфидов щелочных металлов вследствие образования тиосолей. Легче всего идет растворение в случае Au+, для тиосолей которого характерны типы М[АuS] и М3[АuS2]. Выделяющиеся при подкислении их растворов свободные тиокислоты неустойчивы и тотчас распадаются на Аu2S и Н2S.
109) Селениды и теллуриды общих формул Э2Sе и Э2Те известны для всех элементов подгруппы меди. Лучше других изучены производные серебра могут быть получены, в частности, по схеме
4 АgNО3 + 3 Э + 3 Н2О = 2 Аg2Э + Н2ЭО3 + 4 НNО3
взаимодействием селена или теллура (Э) с горячим раствором азотнокислого серебра.
111) При нагревании до 50 °С водного раствора СuSO4 с фосфорноватистой кислотой (Н3РО2) из него медленно выделяется светлый красно-коричневый осадок, в котором соотношение меди и водорода близко отвечает простейшей формуле СuН. Более чистый (и безводный) гидрид меди осаждается из пиридино-тетрагидрофурано-эфирной среды в результате реакции по схеме
4 СuI + LiАlН4 = LiАlI4 + 4 СuН
Для теплоты образования из элементов дается значение -5 кДж/моль. В его кристаллах d(CuH) = 173 и d(CuCl) = 289 пм (против 256 пм у металлической меди). Гидрид меди устойчив на воздухе и по отношению к разбавленным кислотам, но с концентрированной HCl взаимодействует по схеме:
СuН + НСl = Н2 + СuСl
Нагревание (в сухом состоянии выше 60 °С, во влажном — выше 45 °С) вызывает распад СuН на элементы.
114) С азотом элементы подгруппы меди непосредственно не соединяются. Темно-зеленый нитрид меди (Сu3N) является эндотермичным соединением (теплота образования из элементов — 75 кДж/моль) и может быть получен нагреванием СuО до 270 °С в токе аммиака. Он устойчив на воздухе при обычных условиях, но разлагается разбавленными кислотами. Нагревание выше 300 °С ведет к распаду Сu3N на элементы.
115) Белый (на свету чернеющий) осадок амида серебра может быть получен действием КNН2 на раствор соли Аg+ в жидком аммиаке. Как и черный имид Ag2NH, АgNН2 очень взрывчат.
116) При действии аммиака на водную суспензию Аu2О образуется нитридное производное состава Аu3N·NН3, которое после промывания разбавленной кислотой переходит в Аu3N·аq. В сухом состоянии оба соединения взрывчаты.
118) С у г л е р о д о м медь и ее аналоги непосредственно не соединяются. Однако карбиды Сu+, Аg+ и Аu+ могут быть получены косвенным путем — действием ацетилена на аммиачные растворы солей Сu+ и Аg+ или на раствор тиосульфатного комплекса Аu+. Образующиеся карбиды (точнее, ацетилиды) — коричново-красный Сu2С2, белый Аg2С2 и желтый Аu2С2 — в воде практически нерастворимы и в сухом состоянии чрезвычайно взрывчаты. Известен также ацетиленид двухвалентной меди — СuС2.
119) Взаимодействием Ag2С2 с КСєСН в жидком аммиаке был получен комплексный ацетиленид К[Аg(ССН)2]. Он представляет собой бесцветные кристаллы, очень чувствительные к свету и влаге. Известен и близкий по свойствам бесцветный К[Аu(ССН)2].
120) Для золота получено циклопентадиенильное производное — С5Н5Au. Оно представляет собой желтый порошок неустойчивый уже при обычной температуре.
123) По отношению к нагреванию оксид меди довольно устойчив: распад его на Сu2О и кислород начинается лишь около 800 °С (давление кислорода в 1 атм достигается при 1110 °С). Под повышенным давлением кислорода СuО плавится при 1335 °C, а в атмосфере водорода легко восстанавливается уже при 250 °С. Легко восстанавливается он до металла и при прокаливании с углем.
125) В процессе нейтрализации кислых растворов солей, Сu(ОН)2, осаждается около рН = 5. Осаждением раствора СuSO4 щелочью в присутствии (NН4)2SО4 может быть получен кристаллический гидроксид меди (ПР = 2·10-19). Такая его форма начинает отщеплять воду лишь около 150 °С. Электролитическая диссоциация иона СuОН• характеризуется значением К = 3·10-7.
126) В избытке концентрированного раствора сильной щелочи гидроксид меди растворим вследствие образования синих купритов (NаНСuО2, Nа2СuО2 и т. п.). Однако последние весьма неустойчивы и при разбавлении раствора разлагаются с выделением Сu(ОН)2. Это показывает, что кислотные свойства гидроксида меди выражены очень слабо (по приблизительной оценке К1 = 10-10 и К2 = 10-13).
В твердом состоянии из купритов получены лишь производные некоторых щелочных и шелочноземельных металлов. Судя по числу молекул кристаллизационной воды, они имеют комплексную структуру. Например, синему куприту натрия отвечает формула Na2[Cu(OH)4] а светло-синему куприту бария — Ва2[Сu(ОН)6]. Такая трактовка косвенно подтверждается трудностью обезвоживания рассматриваемых соединений. Так, первая из приведенных солей отщепляет воду лишь выше 180 °С, вторая — лишь выше 250 °С.
128) [Сu(NH3)4]2+ представляет собой квадрат с d(СuN) = 205. Его константа устойчивости равна 1·10-13. Помимо различных солей этого комплексного катиона, в виде кристаллогидрата [Сu(NН3)4](ОН)2·3Н2О было выделено и основание. При нагревании его солей до 150-250 °С отщепляется часть аммиака и образуются соответствующие соли катиона [Сu(NН3)2]2+. Вследствие образования аммиачных комплексов металлическая медь при доступе воздуха постепенно растворяется в NН4ОН. Посинение растворов солей меди от добавления аммиака было известно уже Либавию.
130) Для теплот образования галогенидов СuГ2 из простых веществ даются значения 535 (F), 171 (Cl), 134 (Вr) и 8 (I) кДж/моль. В кристаллах белого СuF2 (т. пл. 770 °С) атомы меди имеют шестерную, но неравноценную координацию (4F на расстояниях 193 пм и 2F на расстояниях 227 пм). Безводный СuСl2 (т. пл. 436 °С) окрашен в желтый, а CuBr2 — в черный цвет. Последний легко диссоциирует по схеме
2 СuВr2 = 2 CuВr + Br2
(давление диссоциации в 1 атм достигается уже при 290 °С). Иодид меди (CuI2) не получен. Взаимодействие Сu и I’ сопровождается образованием моноиодида меди (СuI) с одновременным выделением свободного иода по схеме
2 Сu + 4 I’ = 2 СuI + I2
Реакция эта иногда используется для количественного определения меди.
131) Растворимость галогенидов СuГ2 в воде составляет приблизительно 45 (F), 75 (Сl) и 120 (Br) г/л. Из растворов выделяются кристаллогидраты — синий СuF2·2Н2О, зеленый (в присутствии сорбированной воды голубой) СuСl2·2Н2O и коричневато-зеленый СuВr2·2Н2О (или 4Н2О). Обе молекулы воды отщепляются при 132 °С.
138) Малорастворимый в воде (0,08 г/л) коричневый азид Сu(N3)2 может быть получен обменным разложением Сu(NО3)2 с NаN3. Он очень взрывчат (и детонирует в 6 раз сильнее азида свинца). Желтый ион СuN3• малоустойчив.
139) Нитрат двухвалентной меди интересен прежде всего своей летучестью. Металлическая медь реагирует со смесью N2О4 и этилацетата, образуя кристаллический комплекс состава Сu(NО3)2·N2O4, который легко отщепляет N2O4 (давление диссоциации равно 1 атм уже при 85 °С). Остающийся нитрат меди под сильно уменьшенным давлением при 150-200 °С возгоняется и конденсируется на холодной поверхности в виде сине-зеленых кристаллов (т. пл. 226 °С). Давление пара Си(NО3)2 равно 1 мм рт. ст. при 160 °С, а теплота сублимации составляет 67 кДж/моль. В парах Сu(NО3)2 мономерен, причем молекула имеет плоскую структуру с атомом меди, координированным четырьмя атомами кислорода, по два от каждой нитратной группы [d(СuО) = 200 пм, РОСuО = 70°, РОNО = 120°, d(СuN) = 230 пм]. Он хорошо растворим в ряде полярных органических жидкостей, а при нагревании разлагается на СuО, NО2 и О2 (чем иногда пользуются для получения СuО).
144) При взаимодействии Сu•• и СО3” осаждаются труднорастворимые основные карбонаты, встречающиеся в природе в виде очень красивых минералов — зеленого м а л а х и т а [СuСО3·Сu(ОН)2] и синего а з у р и т а [2СuСО3·Сu(ОН)2]. Обработкой основных карбонатов диоксидом углерода под давлением 450 атм при 180 °С был получен нормальный карбонат меди — СuСО3. Известны также некоторые комплексные карбонаты, например голубой К2[Сu(СО3)2]·3Н2О. Благодаря их образованию осадок основных карбонатов Сu2+ растворяется в большом избытке углекислой щелочи.
145) Сулъфат двухвалентной Сu служит обычным исходным продуктом для получения остальных ее соединений. Кристаллогидрат СuSO4·5Н2О (медный купорос) непосредственно применяется для борьбы с вредителями сельского хозяйства, изготовления минеральных красок, в медицине и т. д. Сообщалось также, что добавка его в корм свиней способствует увеличению привеса. Технически медный купорос получают обработкой отходов металлической меди серной кислотой при доступе воздуха.
В сухом воздухе медный купорос частично выветривается и переходит в кристаллогидрат СuSO4·3Н2О, нагревание которого ведет к дальнейшему отщеплению воды, причем сначала образуется СuSO4·Н2О, а затем (выше 258 °С) безводный сульфат меди. Его термическое разложение становится заметным выше 650 °С. Он бесцветен, но на воздухе притягивает влагу и синеет вследствие образования кристаллогидратов.
Растворимость СuSO4 в воде по мере повышения температуры проходит через плоский максимум (рис. Х111-51). В растворе эта соль умеренно диссоциирована (К = 5 ° 10 ') и заметно гидролизована (в 0,1 М растворе при 15 °С степень гидролиза равна 0,05 % и рН = 4,2). С сульфатами щелочных металлов и аммония CuSO4 образует комплексные соли, большей частью отвечающие составу М2[Сu(SO4)2]·6Н2O.
146) Сине-зеленый ацетат двухвалентной меди — Сu(СН3СОO)2·Н2O — в твердом состоянии димерен. Как видно из рис. Х111-52, кажлый атом меди координирован четырьмя атомами кислорода двух ацетатных групп [d(СuО) = 197 пм], одной молекулой воды (d(СuО) = 220 пм] и другим атомом меди [d(СuСu) = 265 пм]. Нагреванием кристаллогидрата при 100 °С в вакууме может быть получен зеленый безводный ацетат. Соль эта растворима не только в воде (25 г/л при обычных условиях), на а в ряде органических жидкостей. Электролитическая диссоциация Сu(СН3СОО)2 характеризуется значениями К1 = 0,12 и К2 = 4·10-3. Комплексные ионы [Сu(СН3СОО)3]’ и [Сu(СН3СОО)4]” малоустойчивы.
147) Бледно-голубой оксалат меди образует почти нерастворимый в воде моногидрат — СuС2О4·Н2О. При нагревании в атмосфере азота он разлагается до металла. Известны и многочисленные комплексные оксалаты меди, главным образом типа М2[Сu(С2O4)2]·2Н2О.
148) Черный сульфид двухвалентной меди (СuS) тотчас образуется при взаимодействии ионов Сu•• и S” в слабокислой среде. Для его произведения растворимости дается значение ПР = 8·10-36. В воде и разбавленных кислотах сульфид этот практически нерастворим. Он слегка растворяется в (NН4)2S и заметно растворим в растворах полисульфидов щелочных металлов и многосернистом аммонии. Во влажном состоянии СuS постепенно окисляется на воздухе с образованием медного купороса.
153) Из простых солей двухвалентного серебра хорошо изучено только AgF2. Фторное серебро представляет собой коричнево-черное (в чистом состоянии бесцветное) вещество плавящееся около 690 °С, которое может быть получено действием фтора на мелкораздробленное Аg (или АgСl). Теплота его образования из элементов равна 347 кДж/моль. Водой AgF2 тотчас разлагается с образованием АgF, НF и кислорода (со значительным содержанием озона). Помимо окислительных оно обладает и сильными фторирующими свойствами (например, способно переводить ССl4 в СF4).
154) Описаны также имеющие коричневые оттенки Аg(VО3)2, АgСО3, АgSiO3, АgСrO4. Сплавлением Ag2О2 с безводной Н3РО4 был получен бесцветный, хорошо растворимый в воде Ag3(PO4)2. Для всех этих веществ известен пока только химический состав, поэтому действительное валентное состояние в них серебра неясно.
Напротив, имеющие голубые оттенки соли типа Аg[ЭF6] (где Э — Sn, Рb, Zr), судя по магнитным данным, действительно являются производными двухвалентного серебра. При хранении на воздухе они разлагаются.
156) Сильные окислительные свойства АgО были использованы для конструирования аккумуляторов типа АgОЅКОНЅZn, работающих по схеме АgО + Zn + 2 КОН + Н2О Ы Аg+ + К2[Zn(ОН)4]. Электролитом служит очень небольшое количество крепкого раствора КОН (с пл. 1,40 г/см3). Такой аккумулятор отличается плотной сборкой и может быть использован при температурах от -50 до +80 °С. Его рабочее напряжение составляет около 1,5 В и весьма постоянно во времени. При равной емкости он в 3 раза меньше и в 5 раз легче свинцового.
157) По литературным данным, при нагревании порошка золота до 140 °С в токе сухого хлора образуется темно-красное соединение, отвечающее эмпирической формуле АuСl2. Описано также несколько других производных Аu (темно-зеленый АuО, красный АuSO4 и т. д.), формально отвечающих двухвалентному золоту. В действительности все они являются, по-видимому, комплексными соединениями, содержащими в своем составе одновременно одно- и трехвалентное золото: АuI[AuIIIСl4], АuI[AuIIIO2] и т. д. С другой стороны, отмечалась неудача попыток получения AuСl2 и было показано, что в системе АuСl-АuСl3 этому составу отвечает не химическое соединен, а эвтектика (около 250 °С). Таким образом, вопрос о реальности существования рассматриваемых производных пока неясен.
159) Аураты щелочных металлов хорошо растворимы в воде, щелочноземельных (а также Мg2+ и Тl+) — умеренно, а большинство остальных — очень мало. При подкислении растворы Na[Au(ОН)4] осаждение Аu(ОН)3 начинается около рН = 7. Производящиеся от НАuО2 аураты могут быть получены сухим путем, например сплавлением золота с селитрой:
Аu + NаNО3 = NаАuО2 + NО
Производные АuIII и многих анионов (NО3-, SO42- и др.) известны только в виде комплексных соединений. Растворы последних обычно содержат очень мало ионов Аu•••.
163) С фтором золото взаимодействует лишь около 400 °С, причем выделить продукты реакции в чистом виде не удается. Нагреванием порошка золота с ВrF3 и последующей отгонкой избытка последнего в вакууме может быть получен оранжевый фторид трехвалентного золота. Для теплоты его образования из элементов дается значение 347 кДж/моль. По кристаллической структуре он представляет собой цепочечный полимер, образованный квадратами из атомов фтора. Внутри каждого такого квадрата находится атом золота, ближе связанный с двумя атомами фтора [d(АuF) = 191 пм], чем с двумя другими [d(АuF) = 2,04 пм], которые являются мостиковыми.
164) Водой AuF3, полностью гидролизуется, а с ВrF3 дает двойное соединение состава АuВrF6 (с вероятной структурой [ВrF2][АuF4]).
165) При действии КСN раствор АuСl3 обесцвечивается вследствие образования [Аu(СN)4]’ (в щелочных средах также ионов [Аu(СN)5” и [Au(CN)6]’”). Упаривание раствора сопровождается выделением кристаллогидрата K[Аu(CN)4]·H2O. Нагревание его до 200 °С ведет к обезвоживанию, а при дальнейшем нагревании происходит отщепление циана с образованием К[Аu(СN)2)]. При действии на последний комплексных галогенидов образуются смешанные галогенидо-цианистые комплексы типа К[Au(СN)2Г2).
167) Свободная золотосинеродистая кислота может быть получена в виде бесцветного кристаллогидрата Н[Аu(СN)4]·3Н2О действием НСN на раствор хлорного золота. Реакция эта интересна как хороший пример полного обращения за счет комплексообразования обычно наблюдаемого направления процессов: очень слабая синильная кислота выделяет в данном случае очень сильную соляную из ее соли. Подобно H[AuCl4], железосинеродистая кислота хорошо растворима в воде, спирте и эфире. При ее прокаливании остается чистое золото.
169) Нитрат и сульфат трехвалентного золота могут существовать только в концентриронанных растворах соответствующих кислот. При разбавлении водой происходит гидролиз с осаждением Аu(ОН)3. Золото в подобных растворах находится, по-видимому, в составе комплексного аниона. Это отчасти доказывается тем, что при медленном упаривании раствора Аu(ОН)3 в концентрированной НNО3 выделяется желтый кристаллогидрат комплексной кислоты Н[Аu(NО3)4]·3Н2О.
170) В противоположность сульфату с е л е н а т трехвалентного золота выделен. Его мелкие желтые кристаллы могут быть получены из раствора Аu в нагретой выше 200 °С безводной селеновой кислоте. Реакция образования этого соединения идет по уравнению:
2 Аu + 6 Н2SеО4 = Аu2(SеО4)3 + 3 SеО2 + 6 Н2О
171) Действием К2SO3 (в присутствии КОН) на НАuСl4 может быть получен бесцветный сульфитный комплекс трехвалентного золота — K5[Au(SO3)4]·5H2O. Интересны известные для ряда катионов кислые соли комплексной кислоты Н7[Аu(IO6)2]. Как правило, они малорастворимы в воде. Важнейшим исключением является Nа6Н[Аu(IO6)2]·16Н2О.
172) Действие Н2S на соединения трехвалентного золота в водной среде ведет на холоду к образованию коричнево-черного осадка Au2S3 по схеме
8 АuCl3 + 9 H2S + 4 Н2О = 4 Аu2S2 + Н2SO4 + 24 НСl
а при нагревании — к восстановлению AuIII до металла. Однако сульфид трехвалентного золота (Аu2S3) может быть получен обработкой сероводородом раствора АuСl3 в безводном эфире. Он представляет собой черный порошок, при нагревании выше 200 °С распадающийся на элементы.
173) Взаимодействие Аu2S3 с раствором Nа2S сопровождается образованием растворимого тиоаурата:
Аu2S3 + Nа2S Ы 2 NаАuS2
Последний был выделен в виде бесцветного кристаллогидрата NаАuS2·4Н2О. Водой этот тиоаурат гидролизуется:
2 NаАuS2 + Н2О Ы Аu2S3 + NаНS + NаОН
По-видимому, он имеет также тенденцию к распаду по схеме: NаАuS2 = NаАuS + S
Был описан и желтый малорастворимый в воде полисульфид состава NН4АuS3.
174) При действии аммиака на соединения трехвалентного золота образуется грязно-желтые осадки переменного состава, в сухом состоянии крайне взрывчатые («гремучее» золото). В присутствии избытка NН4Сl выпадает невзрывчатое амидное соединение — (NН4)2АuСl. Наконец, в присутствии избытка NH4NO3 образуется бесцветный комплексный катион [Аu(NН3)4]•••, для которого кроме азотнокислой известен и ряд других солей.
175) Производные трехвалентных серебра и меди изучены сравнительно мало. Для перехода АgIII + е-Ы АgII в щелочной среде дается значение потенциала +0,74 B. Промежуточное образование Аg••• по схеме
S2O3”+ Аg•= 2 SO4” + Аg•••
является вероятной причиной каталитической активности Аg• при окислении различных веществ персульфатами.
176) Оксид Аg2О3 в индивидуальном состоянии не выделен. Близкие к этому составу осадки образуются, в частности, при пропускании озона сквозь кислый раствор АgNО3. Наиболее чистый Аg2О3 (все же содержавший примесь F-) был получен анодным окислением серебра при электролизе крепкого раствора AgF. этот препарат представлял собой серовато-стальные кристаллы, медленно отщепляющие часть кислорода уже при обычных температурах. При взбалтывании его с большим количеством воды извлекались Аg• и F’, а дебаеграмма остатка содержала линии Аg2О3 и АgО.
177) Ф т о р и д трехвалентного серебра при непосредственном взаимодействии элементов не образуется, но фторированием смеси MСl + АgСl при высоких температурах были получены желтые, очень чувствительные к влаге комплексные соли типа МAgF4 (где М — К, Сs). Их взаимодействие с жидкой НF идет по уравнению
МАgF4 + НF = М++ НF2- + АgF3
и сопровождается осаждением очень неустойчивого аргентотрифторида в виде красно-коричневого порошка. Известен также желтый ВаАgF4 (который при нагревании выше 200 °С в вакууме теряет часть фтора с образованием черного ВаАgF4, устойчивого до 450 °С}. Сообщалось и о получении Сs2К[AgF6] (прямым фторированием смеси 2 СsСl + КCl + АgNО3). Довольно характерны для AgIII кислые соли его комплексов с ортоиодной и ортотеллуровой кислотами, например коричневая К6Н[Аg(IO6)2]·10Н2О и бледно-желтая Nа7Н2[Аg(ТеО6)2]·14Н2О.
178) Оксид трехвалентной меди (Сu2О3) и отвечающий ему гидроксид не получены. Производящиеся от последней к у п р а т ы типа МIСuО2 или МII(СuО2)2 известны для щелочных (Nа-Сs) и щелочноземельных металлов. Общим способом их образования является обработка Сu(ОН)2 в сильно щелочной среде избытком гипохлорита или гипобромита. Реакция идет, например, по уравнению:
2 Сu(ОН)2 + Ва(ОН)2 + NаОСl = Ва(СuО2)2 + NаСl + 3 Н2О
Осадки купратов содержат кристаллизационную воду и имеют различные оттенки красного цвета. Наиболее устойчива из них соль бария, тогда как NаСuО2 разлагается на 10 % за 8 дней уже при обычных температурах. Полученные сухим путем (нагреванием до 400-600 °С смеси СuО + М2О2 в атмосфере кислорода) безводные купраты щелочных металлов имеют серо-черную или серо-синюю окраску.
179) Фторид трехвалентной меди неизвестен, но нагреванием смеси КСl с СuСl2 в атмосфере фтора был получен бледно-зеленый комплекс К3[СuF6]. Водой он разлагается. Добавлением к раствору СuСl2 щелочного раствора NаОСl, а затем подкисленного раствора Nа2Н3IO6 или Nа2Н4ТеО6 были получены малорастворимые в воде Nа7[Сu(IO6)2]·16Н2О и Mа9[Сu(ТеО6)2]·20Н2О. Атом меди иона [Сu([IO6)2]7- находится в центре квадрата из атомов кислорода [d(СuО) = 193 пм], образованного ребрами двух октаэдров [IO6]5-. Известен и ряд кислых солей аналогичных типов. Как правило, они имеют различные оттенки коричневого цвета.
180) Интересно, что как бы промежуточным между Аg+ и ионами щелочных металлов (главным образом К+ и Rb+) является ион одновалентного таллия. Действительно, по ряду свойств Тl+ близко примыкает к Аg+, а по ряду других — к ионам щелочных металлов. Так, подобно гидроксидам последних, ТlOН хорошо растворим в воде и является сильным основанием. Его карбонат (Тl2СО3) также довольно хорошо растворим и по свойствам похож на соду. Тенденция к комплексообразованию с аммиаком в водном растворе у иона Тl+ отсутствует. Многие его соли (Тl2SO4, ТlСlO4 и т. д.) кристаллизуются изоморфно с соответствующими солями К+ и Rb+. Ряд других солей Тl+, напротив, похож на соответствующий ряд солей Аg+. Очень велико, например, сходство между галогенидами обоих элементов, их сульфидами и т. д.
Подгруппа мышьяка.
Содержание элементов этой подгруппы в земной коре сравнительно невелико, и по ряду мышьяк (1·10-4 %) — сурьма (5·10-6 %) — висмут (2·10-6 %) уменьшается. Встречаются они главным образом в виде сернистых минералов — реальгара (As4S4), аурипигмента (As2S3), сурьмяного блеска (Sb2S3) и висмутового блеска (Вi2S3). Примеси всех трёх элементов часто содержатся в рудах различных металлов.
Соединения Аs, Sb и Bi были известны ещё в древнем Египте. Получение элементарного мышьяка из природного сульфида описано в энциклопедии Зосимоса, а при раскопках Вавилона были найдены сосуды из сурьмы, изготовленные за 3000 лет до н. э. Первые упоминания о металлическом висмуте содержатся в алхимических сочинениях ХV века. Мышьяк и висмут — чистые элементы 75Аsи209Вi, тогда как сурьма состоит из двух изотопов — 121 (57,25 %), 122 (42,75 %).
Для получения As, Sb и Bi их сернистые руды обжигаются на воздухе, причём сульфиды переходят в оксиды, которые затем восстанавливают углём. Реакции идут по схемам:
2 Э2S3 + 9 O2 = 6 SO2 + 2 Э2O3 и Э2O3 + 3 C = 3 CO + 2 Э.
В свободном состоянии элементы подгруппы мышьяка имеют металлический вид и довольно хорошо проводят тепло и электричество. Однако они очень хрупки и легко могут быть измельчены в порошок. Важнейшие их константы (наряду с соответствующими данными для азота и фосфора) сопоставлены ниже:
| Элемент | Агрегатное состояние | Цвет | Температура плавления °С | Температура кипения °С | Плотность г/см3 |
| N | газ | бесцветный | -210 | -196 | 1,0 |
| P | твёрд. | белый | 44 | 257 | 1,8 |
| As | твёрд. | серебристый | 817 (36 атм) | 615 | 5,7 |
| Sb | твёрд. | серебристый | 631 | 1634 | 6,7 |
| Bi | твёрд. | красноватый | 271 | 1552 | 9,8 |
На воздухе при обычных условиях Sb не изменяется, а As и Bi слегка окисляются с поверхности. Ни в воде, ни в органических растворителях мышьяк и его аналоги нерастворимы. Со многими металлами они легко дают сплавы.
Обычные формы всех трёх элементов характеризуются однотипной слоистой структурой кристаллов. Каждый атом связан с тремя другими того же слоя и имеет трёх ближайших соседей в другом слое. Сурьма способна образовывать смешанные кристаллы и с Аs, и с Bi, но последние не образуют их друг с другом.
При нагревании (в присутствии воздуха) Аs возгоняется (т. возг. 615 °С). Пар состоит из молекул Аs4 с ничтожной (порядка 0,03%) примесью молекул Аs2. При дальнейшем его нагревании равновесие по схеме Э4 Ы 2Э2 Ы 4Э всё более смещается вправо. То же самое характерно для паров сурьмы и висмута, которые при температурах кипения имеют следующие составы: 49% Sb4 + 49% Sb2 + 2% Sb.
Энергия диссоциации (кДж/моль) двухатомных молекул по ряду N2 (945) - P2 (490) - As2 (385) - Sb2 (300) - Bi2 (200) последовательно уменьшается.
Подобно фосфору, мышьяк способен существовать в нескольких аллотропических формах, из которых наиболее устойчива обычная серая. С повышением давления её температура плавления довольно быстро возрастает (достигая 950 °С при 60 тыс. атм). При очень быстром охлаждении паров получается жёлтый мышьяк с плотностью 2,0 г/см3, довольно хорошо растворимый в сероуглероде (около 8% при 20 °С) и образующий при упаривании такого раствора жёлтые кристаллы. Последние слагаются из молекул Аs4, имеющих, как и у фосфора структуру правильного тетраэдра. На воздухе он легко окисляется, а под действием света быстро переходит в серую форму. При возгонке Аs в струе водорода образуется аморфный чёрный мышьяк с плотностью 4,7 г/см3. Последний не окисляется на воздухе, но выше 270 °С переходит в серую форму.
Элементарный мышьяк используют главным образом в качестве добавки (порядка 0,3%) к свинцу при выработке дроби. Эта добавка повышает твёрдость металла и сообщает ему способность застывать в виде капель строго шарообразной формы. Соединения мышьяка применяются в медицине, при выделке кож и мехов, в стекольном, фарфоровом и других производствах. Важной областью их использования является сельское хозяйство, где различные производные As служат одним из основных средств борьбы с вредителями культурных растений. Ежегодная мировая добыча As составляет около 50 тыс. т.
В отношении аллотропии под обычным давлением сурьма похожа на мышьяк. Её жёлтая форма может быть получена окислением SbH3 озонированным кислородом при -90 °С. Она всегда содержит значительную (порядка 10 атомных %) примесь химически связанного водорода. Повышение температуры сопровождается отщеплением водорода и переходом в чёрную сурьму (с плотностью 5,3 г/см3), которую можно получить и быстрой конденсацией паров Sb. Чёрная форма уже при слабом нагревании переходит в обычную серую. При электролизе сильно охлаждённых концентрированных растворов SbCl3 на катоде осаждается похожая на графит аморфная масса (плотность 5,8 г/см3), содержащая в своём составе значительные количества хлора. Трение вызывает её экзотермический распад с выделением белого дыма. Неустойчивость этой “взрывчатой” сурьмы связана с одновременным наличием в ней структурных элементов и металла (атомов Sb), и соли (ионов SbCl2+, SbCl2+ и Cl-). Под давлением около 85 тыс. атм обычная сурьма переходит в иную аллотропную форму.
Сурьма является важной составной частью некоторых ответственных сплавов (типографский шрифт, сплавы для подшипников и др.). Она применяется также при изготовлении шрапнельных пуль. Добавка к свинцу уже 1% Sb сильно повышает его твёрдость, что имеет большое значение для производства свинцовых труб. Соединения сурьмы используются в резиновой, стекольной, красильной, спичечной и других отраслях промышленности. Ежегодная мировая добыча Sb составляет около 50 тыс. т.
Имеются также указания на возможность получения (нагреванием металла до 110 °С в 70%-ном растворе НСlO4) “взрывчатого” Bi, аналогичного соответствующей форме сурьмы.
Обычная форма висмута обладает некоторыми интересными особенностями. Электропроводность металлического Bi резко изменяется в момент плавления (теплота плавления 11 кДж/моль). Объём висмута при плавлении заметно уменьшается, т. е. он (подобно воде) ведёт себя в этом отношении аномально.
Так как объём висмута при плавлении уменьшается, увеличение внешнего давления понижает точку плавления. Напротив, у “нормально” ведущих себя веществ точка плавления при увеличении внешнего давления должна повышаться.
Висмут служит главным образом для изготовления различных сплавов, которым он обычно сообщает легкоплавкость. Сплавы эти важны для противопожарной арматуры, сигнальных аппаратов, а также широко используются в качестве припоев. Соединения Bi применяются главным образом в медицине, косметике и в стекольной промышленности. Ежегодная мировая добыча Вi составляет около 10 тыс. т.
Легкоплавкие сплавы состоят обычно из Bi, Pb, Sn и Cd с преобладанием висмута. Температуры их плавления сильно зависят от состава. Так, сплав 50% Bi c 25% Pb, 12,5% Sn и 12,5% Cd плавится при 60,5 °С; сплав 50% Вi с 27% Pb, 13% Sn и 10% Cd — при 70 °С и т. д. Иногда применяются и легкоплавкие сплавы без кадмия или с заменой его на ртуть. Например, сплав 50% Bi с 30% Pb и 20% Sn плавится при 92 °С, сплав 36% Bi с 28% Pb, 6% Cd и 30% Hg — при 48 °С. Сплав 53,5% Bi, 41,5% Pb и 5% Hg пригоден для изготовления металлических карандашей, а сплав 20% Bi с 80% Hg хорошо пристаёт к стеклу и применяется иногда для “серебрения” стеклянных поверхностей. Для спаивания стекла с металлом удобно пользоваться сплавом 50% Pb, 37,5% Bi и 12,5% Sn. Сплав 57% Pb с 29% Bi и 14% Hg легко плавится при трении.
В ряду напряжений As, Sb и Bi располагаются между водородом и медью. Поэтому водорода из кислот они не вытесняют, но могут быть переведены в раствор действием окислителей, например, по реакциям:
2 Аs + 5 Cl2 + 8 H2O = 2 H3AsO4 + 10 HCl
Bi + 4 HNO3 = Bi(NO3)3 + NO + 2 H2O.
Растворимые производные всех трёх элементов ядовиты.
Крепкая серная кислота при нагревании переводит мышьяк в As2O3, а сурьму и висмут — в сульфаты Э2(SO4)3. Разбавленная азотная кислота окисляет их соответственно до H3AsO3 и Sb2O3, а концентрированная — до Н3AsO4 и Sb2O5. Висмут растворяется в разбавленной азотной кислоте с образованием Bi(NO3)3, тогда как крепкая кислота его пассивирует. Растворы щелочей сами по себе на рассматриваемые элементы не действуют, но в присутствии кислорода медленно разъедают As и Sb.
Ничтожные количества мышьяка содержатся во всех животных и растениях. Наиболее богаты им морские организмы. Так, ламинария содержит до 0,01% As. Содержание его в человеческом организме составляет около 10-5%.
Очень малые дозы мышьяка стимулируют жизненные процессы, тогда как в более значительных дозах он сильно ядовит. Эта ядовитость мышьяка нашла своё наглядное отражение в его алхимическом символе (змея в спиралевидном состоянии). Острое отравление проявляется не сразу после введения яда. Оно сопровождается появлением болей в животе, рвоты и поноса. Обычным средством первой помощи является питьё молока или приём внутрь свежеприготовленной сильным взбалтыванием MgO с раствором Fe2(SO4)3 взвеси Fe(OH)3 в воде (по чайной ложке через каждые 10 мин). При хронических отравлениях очень малыми дозами As постепенно развиваются расстройства пищеварительного тракта, поражения слизистых оболочек и т. д. Предельно допустимой концентрацией As в воздухе производственных помещений считается 3·10-4 мг/л.
Сурьма обладает сходным с мышьяком, но слабее выраженным ядовитым действием. Токсичность обоих элементов в трёхвалентном состоянии выше, чем в пятивалентном. Висмут значительно менее токсичен и по характеру вызываемого им отравления более похож не на мышьяк, а на ртуть.
При нагревании на воздухе As, Sb и Bi сгорают с образованием оксидов общей формулы Э2О3. Легко соединяются они также с галогенами и серой. Образование определённых соединений с металлами для рассматриваемых элементов менее характерно, чем для азота и фосфора, однако всё же известны некоторые аналогичные нитридам и фосфидам арсениды, антимониды и висмутиды, например Mg3As2, Mg3Sb2 и Mg3Bi2.
Действием на них разбавленных кислот могут быть получены аналогичные аммиаку и фосфину мышьяковистый (“арсин”), сурьмянистый (“стибин”) и висмутистый (“висмутин”) водород общей формулы ЭН3. Реакции идут по схеме:
Mg3Э2 + 6 HCl = 3 MgCl2 + 2 ЭH3.
Так как соединения эти малоустойчивы, больший или меньший распад их на элементы имеет место уже в момент образования и поэтому практически они всегда выделяются в смеси с водородом. Особенно это относится к BiH3, который из-за своей чрезвычайной неустойчивости почти не изучен.
Арсин и стибин представляют собой бесцветные, очень ядовитые газы с чесночным (AsH3) или похожим на сероводородный (SbH3) запахом. Они довольно хорошо растворимы в воде, но химически с ней не взаимодействуют. Характерные для аммиака реакции присоединения не наблюдаются у арсина и стибина. Оба они являются очень сильными восстановителями, например, при нагревании легко разлагаются на элементы, а будучи подожжены на воздухе, сгорают с образованием воды и соответствующих оксидов.
Помимо разложения арсенидов и антимонидов кислотами, арсин и стибин могут быть получены действием водорода в момент выделения на самые разнообразные растворимые соединения мышьяка и сурьмы, например, по реакции:
As2O3 + 6 Zn + 12 HСl = 6 ZnCl2 + 2 AsH3 + 3 H2O.
Молекулы AsH3 и SbH3 имеют структуры треугольных пирамид с углом при вершине соответственно 92° и 91° [d(AsH) = 152, d(SbH) = 171 пм]. Полярность обоих молекул очень мала.
Арсин (т. пл. -117, т. кип. -62 °С) и стибин (т. пл. -94, т. кип. -18 °С) являются эндотермическими соединениями, при обычных условиях они более или менее устойчивы. Термический распад арсина становится заметным около 300 °С. Ещё легче наступает аналогичный распад SbH3, который при нагревании стибина в отсутствие достаточного избытка водорода может иметь взрывной характер. Наконец, BiH3 очень быстро распадается на элементы уже при обычных условиях. Термический распад арсина и стибина используется при глубокой очистке этих соединений.
Растворимость AsH3 и SbH3 в воде составляет приблизительно 1:5 по объёму. В органических растворителях она значительно выше (например, один объём сероуглерода поглощает до 250 объёмов SbH3). Для мышьяковистого водорода известен устойчивый лишь ниже -10 °С кристаллогидрат AsH3·6H2O. Последовательное образование жёлтого AsH(HgCl)2, коричневого As(HgCl)3 и чёрного Hg3As2 при действии арсина на хлорную ртуть используется иногда для его открытия.
В смесях AsH3 c HI или HBr при низких температурах методом инфракрасной спектроскопии было установлено частичное образование ионов арсония (AsH4+).
Мышьяковистый водород является одним из сильнейших неорганических ядов. Отравление им может иметь место, в частности, при всех случаях получения больших количеств водорода взаимодействием цинка или железа с кислотами, если исходные продукты содержат примесь мышьяка (что бывает очень часто) и работа ведётся без соблюдения достаточных мер предосторожности. Опасность усугубляется тем, что первые признаки отравления (озноб, рвота и др.) проявляются обычно лишь спустя несколько часов после вдыхания AsH3. Основным средством первой помощи является свежий воздух при полном покое пострадавшего. Подобно AsH3, но слабее, действует на организм и SbH3. Если смесь обоих гидридов пропускать через разбавленный раствор AgNO3, то мышьяк переходит в раствор (как Н3АsO3), а сурьма — в осадок (как Sb2O3).
Легко протекающий при нагревании распад мышьяковистого водорода на элементы лежит в основе метода открытия мышьяка, которым пользуются при судебно-медицинских и санитарных анализах. Для проведения реакции испытуемый материал обрабатывают цинком и соляной кислотой, пропуская выделившиеся газы сквозь нагретую стеклянную трубку. При наличии As около места нагрева образуется блестящий чёрный налёт (“зеркало”) элементарного мышьяка. Применяемые для определения цинк и соляная кислота должны быть при помощи “холостого” (т. е. выполняемого без испытуемого материала) опыта тщательно проверены на отсутствие примесей мышьяка.
Следует отметить, что сурьма даёт реакцию, аналогичную мышьяку. Природа “зеркала” может быть установлена по его летучести при нагревании или по отношению к раствору NaOCl (в котором As растворяется, а Sb не растворяется). Аналогично сурьме (но лишь в малой степени) может вести себя при этой реакции и висмут.
Действием эфирного раствора SnCl2 на солянокислый раствор AsCl3 может быть получен нерастворимый в воде, щелочах и кислотах коричневый порошок состава As2H2. Вещество это легко окисляется и имеет тенденцию к самопроизвольному распаду на элементы. Имеются указания на возможность получения устойчивого лишь при низких температурах гидрида As2H4.
При пропускании AsH3 в жидкий аммиак, содержащий растворённый металлический калий, жидкость окрашивается в ярко-желтый цвет. После испарения NH3 остаётся аналогичное амиду калия мышьяковистое производное — KAsH2. Его термический распад по схеме: КАsH2 = H2 + KAs идёт лишь выше 80 °С, тогда как NaAsH2 и LiAsH2 разлагаются соответственно уже при 10 и 0 °С. Таким же путём был получен и красно-коричневый KSbH2, менее устойчивый, чем KAsH2.
Сесквиоксиды As, Sb и Bi отвечают общей формуле Э2О3. Они легко образуются при нагревании элементов на воздухе и представляют собой твёрдые вещества белого (As2O3 и Sb2O3) или жёлтого Bi2O3 цвета. Мышьяковистый ангидрид (As2O3) довольно хорошо растворим в воде, тогда как оба других оксида почти нерастворимы.
Теплота образования сесквиоксидов As, Sb и Bi из элементов составляет соответственно 665, 705 и 575 кДж/моль. Для мышьяковистого ангидрида (иногда называемого “белым мышьяком”) кроме октаэдрической модификации (т. пл. 278 °С) известны две другие: устойчивая выше 200 °С моноклинная (т. пл. 314 °С) и устойчивая выше 310 °С стекловидная. Жидкий As2O3 кипит при 461 °С. Растворимость его в воде составляет около 1,2% при 0 °С и 6% при 100 °С. Нагревание Sb2O3 (т. пл. 656, т. кип. 1456 °С) сопровождается изменением его цвета на жёлтый, а нагревание Bi2O3 (т. пл. 825, т. кип. 1890 °С) — изменением цвета на красно-коричневый. Плотности паров сесквиоксидов мышьяка и сурьмы отвечают при 800 °С удвоенным формулам (As4O6 и Sb4O6), выше 1800 °С — простым. По строению они подобны Р4О6. Растворимость As2O3, Sb2O3 и Bi2O3 составляет при обычных условиях соответственно 9·10-2, 3·10-5 и 2·10-8 моль/л Н2О.
Нагреванием Sb2O3 (или Sb2O5) на воздухе может быть получен белый, почти нерастворимый в воде порошок состава Sb2O4. Теплота образования из элементов этого довольно характерного для сурьмы оксида составляет 907 кДж/моль. При сильном прокаливании он отщепляет кислород и переходит вSb2O3. Сплавлением его со щелочами могут быть получены соли типа M2Sb2O5. Как сам оксид Sb2O4, так и производные от него соли содержат в своём составе одновременно трёх- и пятивалентную сурьму и отвечают структурам (SbO)SbO3 и M2(SbO)SbO4. Аналогичное строение (ЭО)ЭО3 имеют оксиды мышьяка и висмута, которые, однако, для них малохарактерны.
Химические свойства гидроксидов Э(ОН)3 по ряду As-Sb-Bi изменяются весьма закономерно. Все они амфотерны, причём у As(OH)3 преобладает кислотный характер, у Sb(OH)3 — основной, а у Bi(OH)3 кислотная функция выражена столь слабо, что обнаруживается лишь по незначительной растворимости этого гидроксида в крепких растворах сильных щелочей. Таким образом, кислотный характер гидроксидов Э(ОН)3 по ряду As-Sb-Bi быстро ослабевает.
Мышьяковистая кислота (Н3AsO3) известна лишь в растворе. Гидроксид сурьмы (сурьмянистая кислота) и Bi(OH)3 представляют собой белые хлопьевидные осадки. Для обоих элементов характерны продукты частичного обезвоживания гидроксидов — SbO(OH) и BiO(OH). Отвечающие им радикалы — SbO (антимонил) и BiO (висмутил) часто входят как таковые в состав солей и играют в них роль одновалентных металлов.
Растворённая часть гидроксидов As и Sb способна диссоциировать одновременно по суммарным схемам:
Э3+ + 3 ОН-Ы Э(ОН)3є Н3ЭО3Ы 3 Н+ + ЭО33-
При добавлении к раствору кислот равновесие смещается влево и образуются соли катионов Э3+, а при добавлении щелочей равновесие смещается вправо и получаются соответственно арсениты или антимониты: соли с катионом ЭО33-. Кислотная диссоциация может протекать также с отщеплением молекулы воды по схеме:
Н3ЭО3Ы H++ ЭО2- + Н2О,
причём получаются соли метамышьяковистой (HAsO2) и метасурьмянистой (НSbO2) кислот. Обе они являются очень слабыми.
В растворе мышьяковистой кислоты имеет место равновесие по схеме:
НAsO2 + H2O Ы H3AsO3,
сильно смещённое влево, т. е. мета-форма резко преобладает над орто-формой. Кислотные свойства НAsO2 выражены весьма слабо (К = 7·10-10), но всё же гораздо сильнее отвечающих диссоциации по схеме:
ОAsOH Ы OАs++ OH-
основных (К = 5·10-15). Последние проявляются образованием AsOHSO4 при растворении As2O3 в 100%-ной серной кислоте и As(HSO4)3 при растворении его в олеуме. Вторая и третья константы кислотной диссоциации H3AsO3 имеют порядок 10-14. Насыщенный раствор As2O3 показывает рН = 5,0 (при 25 °С).
Большинство арсенитов производится от метамышьяковистой кислоты. Важным для химического анализа ортоарсенитом является малорастворимый (ПР = 1·10-17) жёлтый Ag3AsO3. Входящий в состав этой соли ион AsO33- имеет структуру треугольной пирамиды. Для антимонитов щелочных металлов характерны типы M[Sb(OH)4], MSbO2, M2Sb4O7 и M2Sb6O10.
Так как основные свойства гидроксидов Э(ОН)3 по ряду As-Sb-Bi усиливаются, по тому же ряду возрастает и устойчивость солей с катионом Э3+. В частности, соли кислородсодержащих кислот для As3+ в свободном состоянии вообще не выделены, для Sb3+ известны лишь единичные их представители, в то время как бесцветный Bi(NO3)3·5H2O является наиболее обычным соединением висмута. Растворимые производные Bi3+ и Sb3+ легко разлагаются водой с выделением основных солей.
Хотя соли кислородных кислот для Sb3+ не характерны, растворением Sb (или Sb2O3) в горячей концентрированной серной кислоте всё же может быть получен нормальный сульфат сурьмы — Sb2(SO4)3. С небольшим количеством воды соль эта даёт кристаллогидрат; при разбавлении раствора сперва образуется сульфат антимонила (SbO)2SO4, а затем наступает дальнейший гидролиз. Несколько более устойчивы в растворе двойные соли типа М[Sb(SO4)2]. Нормальный нитрат — Sb(NO3)3 — может быть получен взаимодействием SbCl3 с AgNO3 в ацетоне. Под действием уже следов воды он переходит в основной нитрат. Образующийся при нагревании смеси Sb2O3 + P2O5 ортофосфат сурьмы — SbPO4 — обладает высокой термической устойчивостью (не разлагается даже при 1200 °С).
Весьма характерна для сурьмы смешанная виннокислая соль антимонила и калия состава K(SbO)C4H4O6·H2О. Соль эта (“рвотный камень”) легко образуется при кипячении Sb2O3 с раствором кислого виннокислого калия (KHC4H4O6) и представляет собой бесцветные кристаллы, легкорастворимые в воде. Она находит применение в медицине и красильном производстве.
Азотнокислый висмут может быть получен растворением металла в HNO3. После упаривания раствора он выделяется в виде больших бесцветных кристаллов Bi(NO3)3·5H2O. Соль эта хорошо растворима в эфире и ацетоне. При растворении в воде происходит сильный гидролиз с выделением осадка основных солей переменного состава. Нагревание кристаллогидрата сопровождается отщеплением не только воды, но и части азотной кислоты с образованием в остатке нитрата висмутила — (BiO)NO3.
Бесцветные гигроскопичные кристаллы Bi2(SO4)3 (т. пл. 710 °С) могут быть выделены из раствора, получающегося при взаимодействии Bi (или Bi2O3) с концентрированной серной кислотой. Водой сульфат висмута легко гидролизуется. С сернокислыми солями некоторых одновалентных металлов он образует комплексные сульфаты типов M[Bi(SO4)2] и M3[Bi(SO4)3]. Из углекислых солей висмута известно только производное висмутила состава (BiO)2CO3·xH2O, осаждающееся при действии Na2CO3 или (NH4)2CO3 на растворы солей висмута.
Из соединений трёхвалентного мышьяка практически наиболее важен мышьяковистый ангидрид, являющийся основным исходным продуктом для получения остальных производных As. Непосредственно он применяется в стекольной промышленности (для обесцвечивания стекла), как консервирующее средство (в меховой промышленности и т. д.) и в медицине. Небольшие количества As2O3 благотворно действуют на организм человека и животных (а по некоторым данным и растений). Установлено, что добавление As2O3 в корм скоту заметно повышает его рост и работоспособность. Оксид сурьмы (Sb2O3) применяется для получения различных эмалей и глазурей, оксид висмута — при производстве хрусталя. Из солей наибольшее значение имеет основная азотнокислая соль висмутила приблизительного состава (BiO)NO3·BiO(OH), используемая в медицине при желудочных заболеваниях. Соль эта применяется также в косметической промышленности и при изготовлении красок для живописи.
Параллельно с ослаблением кислотных и усилением основных свойств гидроксидов Э(ОН)3 по ряду As-Sb-Bi ослабляются также и восстановительные свойства, т. е. уменьшается тенденция элементов к переходу в соединения их высшей валентности. Мышьяковистая кислота, будучи сильным восстановителем в щелочной среде, в кислой окисляется уже значительно труднее. Сурьмянистая кислота типичным восстановителем не является, хотя окисление её в щелочной среде идёт довольно легко. Напротив, гидроксид висмута может быть окислен только в сильнощелочной среде и наиболее сильными окислителями.
Высшие оксиды As и Bi — мышьяковистый ангидрид (As2O5) и сурьмяный ангидрид (Sb2O5) — могут быть получены осторожным нагреванием их гидратов, образующихся при окислении элементарных As и Sb крепкой азотной кислотой. Мышьяковистый ангидрид представляет собой белую стекловидную массу, расплывающуюся на воздухе. Жёлтый порошок сурьмяного ангидрида очень мало растворим в воде.
Теплота образования As2O5 из элементов составляет 928 кДж/моль. Он диссоциирует на As2O3 и O2 выше 400 °С. Отвечающая As2O5мышьяковая кислота (H3AsO4) может быть получена по реакции:
3 As + 5 HNO3 + 2 H2O = 3 H3AsO4 + 5 NO.
Для мышьяковой кислоты (К1 = 6·10-3, К2 = 1·10-7, К3 = 3·10-12) очень характерна практически нерастворимая в воде шоколадно-бурая соль серебра. Различием цвета Ag3AsO3 и Ag3AsO4 (ПР = 1·10-20) иногда пользуются для установления валентности находящегося в растворе мышьяка. Ион AsO43- имеет структуру тетраэдра с атомом мышьяка в центре. Арсенаты Са и Pb используются для борьбы с вредителями сельского хозяйства.
Мышьяковая кислота выделяется при обычных условиях из раствора в виде кристаллогидрата состава H3AsO4·1/2H2O (т. е. As2O5·4H2O). Отвечающие по составу пиро- и мета-формам мышьяковой кислоты гидраты при его обезвоживании не образуются. Напротив, NaH2AsO4 изменяется при нагревании подобно соответствующему фосфату:
90 135 230 °С
NaH2AsO4® Na2H2As2O7® Na3H2As3O10® (NaAsO3)х
Образующийся в конечном счёте метаарсенат плавится при 615 °С. При низких температурах (около -30 °С) может быть выделен кристаллогидрат As2O5·7H2O, отвечающий по составу кислоте H7[AsO6]. От последней, как и от аналогичного соединения фосфора производится ряд гетерополикислот и их солей, многие из которых при обычных условиях вполне устойчивы. Гидрат As(OH)5 не получен, но известен производящийся от него As(OCH3)5.
Для Sb2O5 определённые гидратные формы не характерны, и белый аморфный осадок xSb2O5·yH2O изменяет свой состав в зависимости от условий выделения. Он может быть получен обезвоживанием своего гидрата при 275 °С. Теплота его образования из элементов составляет 1007 кДж/моль. В воде он почти нерастворим. Кислотные свойства (К1 = 4·10-5) сурьмяной кислоты выражены довольно слабо.
Соли мышьяковой кислоты (мышьяковистокислые или арсенаты) производятся главным образом от ортогидрата (Н3AsO4) и похожи по свойствам на соответствующие фосфаты. Соли сурьмяной кислоты (сурьмянокислые, или антимонаты) производятся обычно от гексагидроксосурьмяной кислоты — H[Sb(OH)6], отвечающей гидратированной мета-форме HSbO3·3H2O. Подобно фосфатам, арсенаты и антимонаты, как правило, бесцветны и труднорастворимы в воде. Производные К и Pb применяются в керамической промышленности. Образование труднорастворимого Na[Sb(OH)6] используется в аналитической химии для открытия натрия (при отсутствии лития и аммония).
При действии сильных окислителей (Сl2 и т. п.) на суспензию гидроксида висмута в концентрированном растворе NaOH или КОН образуются нерастворимые производные пятивалентного висмута, окрашенные в цвета от фиолетового до жёлтого. Состав их более или менее близок к формулам NaBiO3 или KBiO3. Эти висмутаты являются чрезвычайно сильными окислителями. Так, в кислой среде двухвалентный марганец легко окисляется ими до семивалентного.
В чистом состоянии висмутаты имеют жёлтую окраску. Кроме них были получены оранжевые соли состава: Э(BiO3)2·4H2O (где Э — Са или Ba) и чёрный AgBiO3. Имеется также указание на получение (спеканием Li2O и Bi2O3 в атмосфере кислорода) Li3BiO4, Li5BiO5 и Li7BiO6. Существование НBiO3 и Bi2O5 как индивидуальных соединений сомнительно.
Сравнительная окислительно-восстановительная активность элементов в характерных для них трёх- и пятивалентном состояниях может быть выражена следующей схемой:
AsIII SbIII BiIII AsV SbV BiV
![]()
![]()
Усиление восстановительных свойств Усиление окислительных свойств
Окислительные свойства мышьяковой и сурьмяной кислот заметно проявляются лишь в кислой среде, причём первая способна окислить НI до I2, а вторая — даже HСl до Сl2 по обратимым реакциям:
H3AsO4 + 2 HI Ы H3AsO3 + I2 + H2O
H3SbO4 + 5 HCl Ы SbCl3 + Cl2 + 4 H2O.
Производные пятивалентного висмута являются окислителями уже не только в кислой, но и в щелочной среде.
Весьма характерные для As, Sb и Bi сернистые соединения могут быть получены как взаимодействием этих элементов с серой при нагревании, так и обменным разложением в растворах. Полученные сухим путём (а также природные) Bi2S3 и Sb2S3 представляю собой серо-чёрные кристаллические вещества. Из растворов Bi2S3 выделяется в виде коричнево-чёрного, Sb2S3 и Sb2S5 — оранжево-красных, а As2S3 и As2S5 — ярко-жёлтых порошков. Все эти сульфиды нерастворимы в воде и разбавленных кислотах (не являющихся одновременно окислителями). Сульфиды мышьяка нерастворимы и в концентрированной HCl, но крепкая азотная кислота (и царская водка) растворяет их по реакции:
3 As2S5 + 40 HNO3 + 4 H2O = 6 H3AsO4 + 15 H2SO4 + 40 NO.
Сульфиды As, Sb и Bi проявляют некоторую аналогию свойств с оксидами тех же элементов. Подобно тому, как оксиды As и Sb при взаимодействии со щелочами дают соли кислот Н3ЭО3 или Н3ЭО4, сернистые их производные образуют с растворимыми сульфидами металлов соли соответствующих тиокислот (т. е. кислот, в которых кислород замещён на серу), например, по реакциям:
3 (NH4)2S + As2S3 = 2 (NH4)3AsS3 и 3 (NH4)2S + As2S5 = 2 (NH4)3AsS4.
Также протекает процесс и для сульфидов сурьмы. Напротив, Bi2S3 с растворимыми сернистыми солями практически не реагирует. Сульфид этот, следовательно, ведёт себя аналогично почти нерастворимому в щелочах оксиду (Bi2O3).
Соли тиомышьяковистой (H3AsS3), тиомышьяковой (H3AsS4) кислот и соответствующих кислот сурьмы вполне устойчивы. Как правило, они имеют жёлтый или красный цвет. Производные Na, K и NН4 в воде растворимы хорошо, большинство остальных — плохо. Некоторые тиоарсениты и тиоарсенаты применяются для борьбы с вредителями сельского хозяйства.
В отличие от своих солей, свободные тиокислоты неустойчивы и разлагаются на соответствующий сульфид и сероводород, например, по схемам:
2 Н3AsS3 = As2S3Ї + 3 H2S и 2 H3AsS4 = As2S5Ї + 3 H2S.
Поэтому при подкислении раствора тиосоли отвечающий ей сульфид выпадает в осадок по реакции:
2 (NH4)3AsS4 + 6 HCl = 6 NH4Cl + As2S5Ї + 3 H2S.
Образование и распад тиосолей As и Sb имеют большое значение для качественного химического анализа.
При обычных условиях и нагревании в отсутствие воздуха все сульфиды As, Sb и Bi устойчивы. Например, As2S3 плавится при 310 °С и кипит при 723 °С без разложения, As2S5 распадается на As2S3 и S лишь при 500 °С. Молекула трёхсернистого мышьяка отвечает формуле As4S6 [d(AsS) = 225 пм] и построена однотипно с P4O6. As4S4 плавится при 320 °С и кипит при 534 °С. Пятисернистый мышьяк отвечает формуле As4S10 и построен однотипно с P4O10.
Сульфиды мышьяка применяются в кожевенной промышленности (для снятия волоса со шкур), в пиротехнике и производстве минеральных красок; Sb2S3 (т. пл. 560, т. кип. 1160 °С) используется в пиротехнике, спичечном и стекольном производствах, Sb2S5 — в резиновой промышленности (для вулканизации каучука). Помимо упоминавшихся выше сульфидов известны As2S3, Sb2S4, Bi4S4, BiS2. Для селенидов и теллуридов As, Sb, Bi характерен тип Э2Se3 или Э2Те3. Все эти соединения могут быть получены нагреванием смесей соответствующих элементов, взятых в стехиометрических количествах. Теллурид висмута (т. пл. 580, т. кип. 1172 °С) используется в некоторых термоэлектрических устройствах. Его кристаллы имеют слоистую структуру и обнаруживают резко различную электропроводность в направлениях параллельном и перпендикулярном слоям. С повышением давления их температура плавления сперва возрастает (до 610 °С при 15 тыс. атм), а затем понижается (до 535 °С при 50 тыс. атм).
Так как основной характер As(OH)3 и Sb(OH)3 выражен значительно слабее, чем у гидроксида висмута, осаждение трёхвалентных As и Sb сероводородом нужно вести в кислой среде, для того чтобы сместить равновесие диссоциации обоих гидроксидов в сторону образования катионов Э3+. Ещё более это относится к осаждению сульфидов пятивалентных As и Sb, так как при нейтральной (и тем более щелочной) реакции раствора, в нём содержится лишь ничтожное количество ионов As5+ и Sb5+. Только при большом избытке кислоты (особенно в случае As) равновесие обратимой реакции ЭО43+ + 8 Н+Ы 4 Н2О + Э5+ смещается вправо. Этого достаточно для того, чтобы мог образоваться осадок сульфида Э2S5. При этом наряду с осаждением Э2S5 всегда идёт также окисление сероводородом по схеме: Э5+ + Н2S = Э3+ + S + 2 H+. В результате при осаждении сероводородом производных пятивалентных As и Sb в кислой (обычно солянокислой) среде образуется смесь сульфидов Э2S5 и Э2S3, причём осадок содержит также выделившуюся при окислении серу. В случае Sb восстановление до Э3+ идёт практически нацело, а в случае As состав осадка сильно зависит от условий осаждения.
Весьма вероятно, что промежуточной стадией при осаждении сульфидов пятивалентных мышьяка и сурьмы является образование их тиокислот. С этой точки зрения основные протекающие при осаждении сульфидов процессы выражаются следующими суммарными схемами: ЭО43- + 4 Н2S Ы 4 Н2О + ЭS43-, затем 3 Н+ + ЭS43-Ы Н3ЭS4 и, наконец, 2 Н3ЭS4® Э2S5Ї + 3 Н2S или 2 Н3ЭS4® Э2S3Ї + 2 SЇ + 3 Н2S.
Кроме продуктов полного замещения кислорода на серу, для мышьяка и сурьмы были получены соли многих промежуточных тиокислот. Например, для мышьяковой кислоты известны производные всех членов следующего ряда: H3AsO4, H3AsSO3, H3AsS2O2, H3AsS3O, H3AsS4. Образованием подобных веществ обусловлена растворимость сульфидов As и Sb в щелочах. Ион AsS43- представляет собой тетраэдр с атомом мышьяка в центре и d(AsS) = 223 пм.
Практическое значение для очистки различных газов от H2S и извлечения содержащейся в нём серы имеют реакции:
2 Na2HAsS2O2 + 2 H2S = 2 Na2HAsS3O + 2 H2O и затем
2 Na2HAsS3O + O2 = 2 Na2HAsS2O2 + 2 SЇ.
По первой из них сероводород улавливается, а по второй (осуществляемой продуванием тока воздуха) исходный раствор регенерируется.
Галогениды As, Sb и Bi легко образуются при прямом взаимодействии элементов. Галогениды типа ЭГ3 известны для всех рассматриваемых элементов и галогенов, тогда как из представителей типа ЭГ5 более или менее устойчивы лишь производные фтора и SbCl5.
Практически приходится иметь дело почти исключительно с хлоридами элементов. При обычных условиях AsCl3 иSbCl5 — вещества жидкие, аSbCl5 иBiCl3 — твёрдые. Все четыре хлорида бесцветны и хорошо растворимы в воде, но подвергаются сильному гидролизу. С хлоридами некоторых одновалентных металлов они способны образовывать комплексные соединения, главным образом типов M[ЭCl4] и M[SbCl6].
Получать AsCl3 удобно, пропуская ток сухого HCl над нагретым до 180-200 °С мышьяковистым ангидридом, а SbCl3 — растворяя мелкорастёртую Sb2S5 в горячей концентрированной HСl. Взаимодействие SbCl3 с концентрированной серной кислотой идёт по уравнению:
2 SbCl3 + 3 H2SO4 = Sb2(SO4)3 + 6 HСl.
Для получения BiCl3 либо растворяют в HCl гидроксид висмута, либо обрабатывают висмут царской водкой. Остаток после упаривания подвергают затем перегонке в отсутствие воздуха. Интересно, что под действием света BiCl3 постепенно темнеет, а в темноте вновь обесцвечивается.
Молекулы галогенидов ЭГ3 имеют структуры треугольных пирамид с атомом Э в вершине и углом ГЭГ при ней около 100°. Некоторые свойства рассматриваемых соединений приведены в даваемом ниже сопоставлении:
| Теплота образования, кДж/моль | d(Э-Г), пм | Энергия связи, кДж/моль | Температура плавления, °С | Температура кипения, °С | |
AsF3 | 957 | 171 | 481 | -6 | 58 |
AsCl3 | 313 | 216 | 305 | -16 | 130 |
AsBr3 | 201 | 233 | 251 | 31 | 221 |
AsI3 | 67 | 256 | 192 | 141 | 371 |
SbF3 | 924 | 203 | 435 | 290 | 319 |
SbCl3 | 380 | 233 | 309 | 73 | 233 |
SbBr3 | 259 | 249 | 255 | 97 | 289 |
SbI3 | 100 | 272 | 188 | 171 | 402 |
BiF3 | 899 | 380 | 650 | 900 | |
BiCl3 | 380 | 248 | 280 | 234 | 439 |
BiBr3 | 259 | 234 | 219 | 461 | |
BiI3 | 109 | 180 | 408 | 542 |
Окрашенными из рассматриваемых соединений являются только жёлтый BiBr3, красные AsI3, SbI3 и чёрный BiI3 (для SbI3 известна и менее устойчивая жёлтая модификация).
Молекула AsF3 имеет РFAsF = 95,5° и весьма полярна (m = 2,81). С фторидами Cs, Rb и K (но не Na или Li) арсентрифторид способен образовывать комплексы типа MAsF4. Растворимость SbF3 в воде исключительно велика (4:1 по массе). Висмуттрифторид практически нерастворим в воде, но заметно растворяется в крепких растворах KF или NH4F с образованием комплексных солей типа M[BiF4]. Комплексообразование с солями одновалентных металлов характерно и для SbF3. Наиболее обычным типом образующихся комплексов является M[SbF4]. Известны также соли типов M2[SbF5] и M2[Sb2F7]. Ион [SbF5]2- имеет структуру октаэдра, в котором одно из направлений отвечает свободной электронной паре центрального атома, а в ионе [Sb2F7]2- два таких октаэдра связаны одним общим атомом фтора.
Арсентрихлорид в ничтожной степени самодиссоциирован по схеме: AsCl3 + AsCl3Ы AsCl2+ + AsCl4-. Он хорошо растворяет серу и фосфор. Будучи хлорангидридом мышьяковистой кислоты, AsCl3 разлагается водой по суммарному уравнению: AsCl3 + 3 H2O Ы As(OH)3 + 3 HCl. В отличие от гидролиза PCl3, реакция эта заметно обратима, и добавлением избытка концентрированной HСl равновесие её может быть смещено влево. Из-за летучести хлористого мышьяка оно смещается в ту же сторону и при кипячении раствора. Из подкисленных водных растворов AsCl3 может быть извлечён эфиром.
Так как основные свойства Sb(OH)3 и Bi(OH)3 выражены значительно сильнее, чем у As(OH)3, гидролиз SbCl3 и BiCl3 протекает с образованием не свободного основания, а основных солей по схемам:
ЭCI3 + H2O Ы Э(OH)Cl2 + HСl и Э(OH)Сl2 + H2O Ы Э(OH)2Cl + HСl.
Образующиеся основные соли типа Э(OH)2Cl легко отщепляют молекулу воды, переходя в нерастворимые хлориды соответственно антимонила или висмутила (хлороксиды Sb и Bi). Поэтому взаимодействие SbCl3 и BiCl3 с водой практически протекает по схеме:
ЭCl3 + H2O Ы ЭOСlЇ + 2 HСl.
Аналогичный по составу хлороксид мышьяка (AsOCl) может быть получен взаимодействием AsCl3 с As2O3. Расплавленный SbCl3 хорошо растворяет многие неорганические соединения. То же относится к SbBr3, тогда как AsBr3 растворяет лишь соединения ковалентного типа. Подобно хлоридам, бромиды и иодиды способны образовывать комплексы с соответствующими солями одновалентных металлов. В частности, с производными типа M[BiI4] приходится встречаться в аналитической практике. Водой бромиды и иодиды разлагаются аналогично хлоридам. Для скоростей гидролиза галогенидов мышьяка было найдено соотношение AsF3 » AsCl3 > AsBr3 > AsI3.
Пентафториды бесцветны и обладают следующими свойствами:
AsF5 | SbF5 | BiF5 | |
| Состояние при обычных условиях | газ | жидкость | твёрдое |
Температура плавления °С | -80 | +8 | 151 |
Температура кипения °С | -53 | 143 | 230 |
Все три фторида являются окислителями, причём по ряду As-Sb-Bi их окислительная активность быстро увеличивается.
Теплота образования AsF5 из элементов равна 1237 кДж/моль, а энергия связи AsF в нём 385 кДж/моль. BiF5 возгоняется в виде белых игольчатых кристаллов при нагревании BiF3 до 600 °С в токе фтора. Соединение это обладает сильным фторирующим действием, бурно реагирует с водой, а во влажном воздухе желтеет и затем белеет вследствие гидролиза. Последний характерен также для фторида мышьяка, тогда как фторид сурьмы гидролизуется значительно меньше соответствующего хлорида.
Для антимонпентафторида известен твёрдый кристаллогидрат SbF5·2H2O, являющийся, вероятно, пентафторгидроксоантимонатом оксония —[H3O][SbF5OH]. Некоторые отвечающие ему по составу соли типа MI[F5OH] были получены и для Sb, и для As. Термическим разложением в вакууме фторонитратов AsF3(ONO2)2, SbF3(ONO2)2 и SbF(ONO2)4 (получаемых взаимодействием соответствующих фторохлоридов с ClNO3) были получены оксофториды AsOF3, SbOF3 и SbO2F, представляющие собой белые гигроскопичные рентгеноаморфные порошки. По-видимому, они полимерны, так как прямое фторирование эквимолярной смеси As2O3 и AsCl3 приводит к образованию мономерного OAsF3, который при обычных условиях газообразен (т. пл. -68, т. кип. -26 °С).
Пентахлорид сурьмы может быть получен по реакции:
SbCl3 + Cl2 = SbCl5 + 54 кДж/моль
Она представляет собой бесцветную жидкость (т. пл. 3, т. кип. 140 °С с частичным отщеплением хлора), под уменьшенным давлением перегоняющуюся без разложения. С хлоридами ряда одновалентных металлов SbCl5 образует довольно устойчивые комплексные соли типа M[SbCl6]. Будучи хлорангидридом сурьмяной кислоты, пентахлорид сурьмы разлагается водой по схеме:
SbCl5 + 4 H2O = H3AsO4 + 5 HCl.
Реакция эта (во избежание восстановления сурьмы проводимая с водой, насыщенной хлором) является удобным методом получения чистой сурьмяной кислоты. Пентахлорид сурьмы применяется в качестве легко отдающего хлор вещества при органических синтезах. По строению молекулы она подобна пятихлористому фосфору.
При смешении бесцветных SbCl3 и SbCl5 образуется тёмно-коричневая жидкость, в которой имеет место сильно смещённое влево равновесие:
SbCl3 + SbCl5Ы 2 SbCl4.
В присутствии CsCl или RbCl выделяются чёрно-фиолетовые кристаллы комплексных солей типа M2[SbCl6]. Известны и бромиды аналогичного типа. Исследования этих солей показало, что они производятся не от четырёхвалентной сурьмы, а содержат равное число атомов SbIII и SbV, т. е. правильнее описываются формулой вида M3[SbCl6]·M[SbCl6]. В растворе они легко распадаются на соответствующие производные трёх- и пятивалентной сурьмы.
Аналогичный SbCl5 пятихлористый мышьяк при взаимодействии AsCl3 с хлором не образуется. Однако известны оранжевые комплексы [PCl4][AsCl6] и [N(C2H5)4][AsCl6] (т. пл. 147 °С с разл.), содержащие связанный с хлором пятивалентный мышьяк в анионе. Вместе с тем из AsF3 и хлора (в присутствии следов воды) может быть получен [AsCl4][AsF6], содержащий связанный с хлором пятивалентный мышьяк в катионе. Известны и некоторые другие производные катиона [AsCl4]+, например[AsCl4]SbCl6 (т. пл. 127 °С). Катион [AsCl4]+легко гидролизуется, тогда как анион [AsF6]- по отношению к воде весьма устойчив. Для аналогичного катиона [SbCl4]+ были получены бесцветные кристаллические производные [SbCl4][SbF6] (по другим данным, SbF3Cl2 с бипирамидным строением), [SbCl4]2SO4 и [SbCl4]F. У последнего соединения, в отличие от [PCl4]F, известна лишь одна форма, в твёрдом состоянии тетрамерная (т. пл. 83 °С).
Бромиды и иодиды ЭГ5 в свободном состоянии не получены. В виде комплексных солей типа M[SbBr6] (и отвечающей им кислоты состава HSbBr6·3H2O) известен бромид пятивалентной сурьмы. Для её иодида получена лишь соль Cs[SbI6].
Для всех рассмотренных выше галогенидов As, Sb и Bi характерна склонность к реакциям присоединения. Проявляется она по отношению к самым разнообразным веществам. Например, известны продукты состава AsCl3·4NH3, BiCl3·NO, BiCl3·NO2, BiCl3·NOCl; SbCl5·NOCl, AsF3·SCl4, AsF5·IF7, SbF5·NO2, SbF5·SO2, SbF5·SF4, SbCl5·ICl3, SbCl5·POCl3 и т. д. Некоторые из этих продуктов присоединения весьма устойчивы. Так, соединение состава SbCl5·6NH3 может быть даже возогнано.
Длительным нагреванием смеси As + I2 в запаянной трубке до 240 °С были получены красные кристаллы As2I4 (т. пл. 136 °С). На воздухе они очень легко разлагаются. Аналогичные производные других галогенидов не получены. Сурьма несколько растворима в расплавленном SbI3, что связано, по-видимому, с частичным образованием нестойкого Sb2I4.
Для висмута описано получение (тремя различными способами) чёрного Bi2Cl4 (т. пл. 163 °С). Однако более поздние исследования говорят о том, что в системе BiГ3-Bi существует только один субгалогенид — BiГ (предположительно в форме BinГn где n = 2,3 или 4). Чёрный хлорид устойчив на воздухе, но выше 320 °С распадается на BiCl3 и Bi. Его образованием обусловлена, по-видимому, хорошая растворимость Bi в расплавленном BiCl3.
Для сурьмы и висмута известны соответствующие солям антимонила и висмутила тиосоединения: красно-коричневый хлористый тиоантимонил (SbSCl), серый хлористый тиовисмутил (BiSCl) и т. д. Эти очень устойчивые по отношению к воде вещества могут быть получены действием газообразного сероводорода на соответствующий галогенид ЭГ3, например, по реакции:
BiCl3 + H2S = 2 HСl + BiSCl.
Сероводород в этом случае реагирует аналогично воде. Подобным же образом при взаимодействии SbCl5 и H2S получается бесцветный тиохлорид SbSCl3.
Производных Sb и Bi типа ЭSГ (где Г = Cl, Br, I) получают путём нагревания смеси соответствующих галогенидов в отсутствии воздуха. Для сурьмы таким образом были получены также SbSeBr, SbSeI и SbTeI, для висмута — BiSeГ (где Г — Cl, Br, I) и BiTeГ (где Г — Br, I).
Нитриды для As, Sb и Bi не характерны. Они образуются в результате разложения соответствующих амидов по суммарной схеме:
Э(NH2)3® 2 NH3 + ЭN,
первоначально возникающих при взаимодействии галогенидов ЭГ3 с жидким аммиаком (или раствором КNH2 в нём). Оранжево-красный AsN разлагается на элементы около 300 °С. Оранжевый SbN начинает разлагаться при 550 °С и очень чувствителен к влаге. Последнее относится и к чёрно-коричневому BiN, который значительно менее устойчив и способен разлагаться со взрывом.
По отношению к фосфору элементы подгруппы мышьяка ведут себя различно. Расплавленные As и P смешиваются в любых соотношениях, тогда как Sb и Bi почти не растворяются друг в друге. При охлаждении расплавов As-P, содержащих от 26 до 47 атомн. % фосфора, в форме чёрных графитоподобных листочков выделяется кристаллическая фаза переменного состава, имеющая структуру типа чёрного фосфора. Взаимодействием AsCl3 с PH3 (при -18 °С) может быть получено соединение обоих элементов состава AsP.
11
Химическое равновесие.
Если смешать газообразные водород и кислород, то взаимодействие между ними в обычных условиях не происходит. Заметные количества воды (водяного пара) начинают очень медленно образовываться лишь примерно с 400 °С. Дальнейшее нагревание исходной смеси настолько ускоряет процесс соединения, что выше 600 °С реакция протекает со взрывом, т. е. моментально.
Таким образом, скорость реакции образования воды из элементов сильно зависит от внешних условий. Для возможности количественного изучения этой зависимости необходимо прежде всего уточнить сами единицы измерения. Скорость химической реакции характеризуется изменением концентрации реагирующих веществ (или продуктов реакции) за единицу времени. Концентрацию чаще всего выражают числом молей в литре, время — секундами, минутами и т. д., в зависимости от скорости данной реакции.
При изучении любого объекта мы всегда так или иначе отделяем его от окружающего пространства. Вещество или смесь веществ в определённом ограниченном объёме (например, в объёме сосуда) называютхимической системой, а отдельные образующие данную систему вещества носят названиееё компонентов. Далее предполагается, что рассматриваемая система представляет собой газ или раствор.
Молекулы той или иной системы могут взаимодействовать лишь при столкновениях. Чем чаще они будут происходить, тем быстрее пойдёт реакция. Но число столкновений в первую очередь зависит от концентраций реагирующих веществ: чем они значительнее, тем больше и столкновений. Наглядным примером, иллюстрирующим влияние концентрации, может служить резко различная энергичность сгорания веществ в воздухе (около 20% кислорода) и в чистом кислороде.
Общую формулировку влияния концентрации на скорость химической реакции даёт закон действующих масс: скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ. Так, для реакции А + В = С имеем u= k[A][B], где u — скорость; k — коэффициент пропорциональности (константа скорости); [A] и [B] — концентрации веществ А и В. Если во взаимодействие вступают сразу несколько частиц какого-либо из веществ, то его концентрация должна быть возведена в степень с показателем, равным числу частиц, входящему в уравнение реакции. Например, выражение для скорости реакции по схеме:
2 Н2 + О2 = 2 Н2О будет: u = k [H2]2[O2].
Близкие к закону действия масс идеи содержались уже в работах Бертолле. Он не смог их обобщить и правильно выразить, так как в то время неясна была разница между концентрацией и общим количеством вещества. В результате поражения Бертолле в полемике с Прустом, как это часто бывает, вместе со всем неверным в его идеях было отвергнуто и всё верное. Из-за этого закон действия масс и вошёл в науку сравнительно поздно. В его разработке участвовал ряд исследователей и современная формулировка этого закона складывалась постепенно.
Закон действия масс может быть выведен на основе следующего положения теории вероятностей: вероятность одновременного осуществления независимых событий равна произведению вероятностей каждого из них. Для того, чтобы произошло химическое взаимодействие, необходимо столкновение реагирующих молекул, т. е. одновременное нахождение их в данной точке пространства. Вероятность (w) такого нахождения для молекулы каждого из веществ прямо пропорциональна его концентрации, т. е. wA = a[A], wB = b[B] и т. д., где a и b — коэффициенты пропорциональности. Отсюда общее число столкновений за единицу времени u = wA·wB = a·b·[A]·[B]… Но успешными, приводящими к химическому взаимодействию, будут не все такие столкновения, а лишь некоторая их доля (a), величина которой при данных внешних условиях зависит только от природы реагирующих веществ. Поэтому скорость реакции u = a·u = a·а[A]·b[B]… Объединяя все константы в одну, получаем закон действия масс. Числовое значение константы скорости (k) выражает скорость реакции в тот момент, когда произведение концентраций реагирующих веществ равно единице.
Возможность осуществления химической реакции должна быть, вообще говоря, тем большей, чем меньшее число отдельный частиц в ней участвует. Это число частиц определяет молекулярность реакции. Так, реакция, сводящаяся к самопроизвольному распаду одной молекулы, является мономолекулярной, обусловленная столкновением двух частиц — бимолекулярной, трёх частиц — тримолекулярной и т. д. Мономолекулярные реакции сравнительно редки. Напротив, бимолекулярные представляют наиболее частый случай. Тримолекулярные реакции уже гораздо более редки, а тетрамолекулярные практически не встречаются.
Действительная молекулярность реакции далеко не всегда совпадает с кажущейся молекулярностью, которая вытекает из суммарного уравнения реакции. “Эмпирические уравнения процессов стоят приблизительно в таком отношении к истинному течению реакций, как эмпирические формулы органических соединений к их конституционным формулам” (Н. А. Шилов). Расхождения между действительной и кажущейся молекулярностями могут иметь место во всех случаях, когда процесс протекает не непосредственно по суммарному уравнению реакции, а через промежуточные стадии. Ход всего процесса определяется в подобных случаях его самой медленной стадией.
Так, около 500 °С с измеримой скоростью идёт формально пятимолекулярная реакция:
4 HBr + O2 = 2 H2O + 2 Br2.
Между тем опыт показывает, что она бимолекулярна. В действительности имеют место следующие стадии:
HBr + O2 = HOOBr медленная стадия
HOOBr + HBr = 2 HOBr быстрая
2 (HOBr + HBr = H2O + Br2) быстрая.
Таким образом экспериментальное определение хода реакции во времени даёт возможность установить её действительную молекулярность и делать важные выводы по вопросу о химизме изучаемого процесса. Однако не следует забывать, что “вывод микроскопического механизма из макроскопических данных никогда не бывает вполне однозначным, и за одной и той же суммарной картиной могут скрываться разные элементарные механизмы” (С.З. Рогинский).
Если в газовых смесях или растворах столкнуться могут любые частицы, то иначе обстоит дело при химических процессах, протекающих с участием твёрдого вещества, потому что в реакцию могут вступать только частицы его поверхности. Поэтому и скорость процесса зависит в данном случае не от объёмной концентрации, а от величины поверхности. Условия для протекания реакции будут, следовательно, тем более благоприятны, чем сильнее измельчено твёрдое вещество.
Кроме концентраций реагирующих веществ на скорость реакции должна влиять температура, так как при её повышении возрастает скорость движения молекул, в связи с чем увеличивается и число столкновений между ними.
Опыт показывает, что при повышении температуры на каждые 10 градусов скорость большинства реакций увеличивается примерно в три раза. Между тем, согласно кинетической теории увеличение числа столкновений при повышении температуры очень невелико и совершенно не соответствует подобным ускорениям реакций.
Число, характеризующее ускорение реакции при нагревании на 10 град, часто называют её температурным коэффициентом скорости. Для подавляющего большинства реакций значения этих коэффициентов при обычных условиях находится в пределах 2-4. По мере повышения температуры они постепенно уменьшаются, приближаясь к единице.
Если исходить из среднего значения температурного коэффициента скорости (3), то нагревание от некоторой начальной температуры (tн) до некоторой конечной (tк) вызывает ускорение реакции в 3w раз, где W = (tк- tн)/10. Например, при нагревании на 100 град она ускоряется в 59 тыс. раз. Между тем число столкновений молекул за единицу времени растёт пропорционально ![]() , где Т — абсолютная температура. Если нагревание производилось, например, от 0 до 100 °С, то число столкновений возрастает всего в
, где Т — абсолютная температура. Если нагревание производилось, например, от 0 до 100 °С, то число столкновений возрастает всего в ![]() :
:![]() = 1,2 раза.
= 1,2 раза.
Это расхождение теории и опыта является, однако, лишь кажущимся. Действительно, химическая реакция не обязательно должна происходить при каждом столкновении частиц реагирующих веществ — может быть очень много таких встреч, после которых молекулы расходятся неизменными. Лишь тогда, когда взаимное расположение частиц в момент столкновения благоприятно для реакции и сталкиваются молекулы достаточно активные, т. е. обладающие большим запасом энергии, они вступают в химическое взаимодействие.
Относительное число подобных “успешных” встреч в первую очередь определяется природой самих реагирующих частиц. Поэтому при одинаковом общем числе столкновений молекул скорости отдельных реакций могут быть весьма различны. С другой стороны, при повышении температуры не только растёт общее число столкновений, но резко возрастает и доля успешных — поэтому так быстро увеличиваются скорости реакций при нагревании. Для различных веществ число активных молекул возрастает при этом в неодинаковой степени — отсюда различия в ускорениях отдельных реакций.
Средняя кинетическая энергия молекул приблизительно равна 0,01 Т кДж/моль, где Т — абсолютная температура. Для обычных условий она составляет около 2,5 кДж/моль, а продолжительность соприкосновения молекул при столкновениях оценивается величинами порядка 10-12 с. За столь короткое время молекулы успевают прореагировать лишь при наличии особо благоприятных условий. Однако общее число столкновений так велико, что даже при одном столкновении из миллиарда (т. е. при a = 10-9) бимолекулярная реакция протекала бы почти мгновенно. Следует отметить, что число столкновений молекул в растворе значительно больше, чем в газе с той же их концентрацией.
Важным условием возможности осуществления химической реакции является подходящее взаимное расположение молекул в момент столкновения. Например, взаимодействию молекул Н-Н и I-I благоприятствует их сближение при параллельности валентных связей. Относительная вероятность возникновения благоприятного для той или иной реакции пространственного расположения молекул оценивается стерическим фактором (числовое значение которого входит в величину a). Например, для рассматриваемой реакции этот фактор близок к 0,1, т. е. благоприятное расположение молекул Н2 и I2 возникает в среднем лишь при одном их столкновении на каждые десять.
Другим важным условием возможности осуществления химической реакции является достаточная реакционная способность молекул в момент столкновения. Особенно реакционноспособными, активными молекулами могут быть наиболее “быстрые”, обладающие значительной кинетической энергией. Ими могут быть также молекулы возбуждённые, у которых некоторые электроны находятся не на нормальном, а на каком-либо более высоком энергетическом уровне. Наконец, активными могут быть молекулы, внутреннее строение которых (расстояние между атомными ядрами и т. д.) в момент столкновения отличается от наиболее устойчивого. Во всех случаях избыточная энергия молекулы обусловливает её повышенную химическую активность.
Энергия необходимая для активирования исходных частиц, носит название энергии активации соответствующей реакции. В начале реакции затрачивается энергия на перевод начальных продуктов (Н) в активное состояние (А) энергия полностью или частично вновь выделяется при переходе к конечным продуктам (К). Поэтому определяемая разностью энергий начальных и конечных продуктов теплота реакции от энергии активации не зависит. Вместе с тем весьма различная в отдельных случаях энергия активации является основным фактором, определяющим скорость реакции: чем больше эта энергия, тем меньше молекул обладают ею при данной температуре и тем медленнее идёт реакция. Как правило, процессы с энергиями активации менее 42 кДж/моль протекают при обычных температурах неизмеримо быстро, а с энергиями активации более 125 кДж/моль — неизмеримо медленно.
От величины энергии активации также зависит температурный коэффициент скорости. Значениям его 2, 3 и 4 при обычной температурах соответствуют энергии активации 59, 88 и 117 кДж/моль.
С точки зрения механизма молекулярного взаимодействия, энергия активации необходима для возбуждения переходного состояния реагирующей системы. Процесс синтеза НI проходит через промежуточное образование “активного комплекса”, в котором исходные связи (Н-Н и I-I) уже расслаблены, а конечные (Н-I) ещё не вполне сформированы. Энергия активации рассматриваемой реакции равна 163 кДж/моль.
Если исходные вещества могут одновременно взаимодействовать друг с другом по двум (или более) различным направлениям, то такие реакции называются параллельными. Из них преимущественно протекает та, которая требует наименьшей энергии активации. Следует подчеркнуть, что даже небольшие различия в этой энергии сказываются на относительных скоростях параллельных реакций очень сильно.
Параллельные реакции гораздо более распространены, чем то кажется на первый взгляд. Лишь сравнительно немногие химические процессы протекают “чисто”, т. е. практически полностью по одному определённому уравнению. Такие реакции особенно ценны для аналитической химии.
В подавляющем большинстве случаев уравнение реакции описывает лишь основной (т. е. количественно преобладающий) процесс. Одновременно протекающие “побочные” реакции обычно не оговариваются, однако считаться с их возможностью приходится почти всегда. По отношению к основному процессу они могут быть либо параллельными (т. е. непосредственно от него не зависящими), либо последовательными (т. е. дальнейшими реакциями продуктов основного процесса).
Если при температурах около 1000 °С водород и кислород со взрывом соединяются, образуя воду, то, наоборот, при 5000 °С вода со взрывом распадается на водород и кислород. Обозначая это схематически, имеем:
———при 1000 °С————Ю
водород + кислород = вода
Ь——при 5000 °С—————
Очевидно, что при некоторых промежуточных температурах должны быть возможны обе реакции. Это действительно имеет место в интервале 2000-4000 °С, когда одновременно происходит и образование молекул воды из водорода и кислорода, и распад молекул воды на водород и кислород. При этих условиях реакция взаимодействия водорода с кислородом становится, следовательно, заметно обратимой. Вообще,обратимыми называютсяреакции, протекающие одновременно в обоих противоположных направлениях. При их записи вместо знака равенства часто пользуются противоположно направленными стрелками.
2 Н2 + О2Ы 2 Н2О
Для скоростей обеих отвечающих данной схеме взаимно противоположных реакций можно составить следующие выражения:
u1 = k1[H2]2[O2] и u2 = k2[H2O]2
Если u1 > u2, то за единицу времени молекул воды будет образовываться больше, чем распадаться. Если же u1 = u2, то число распадающихся и образующихся за единицу времени молекул воды будет одинаково.
Допустим, что водяной пар нагрет до 3000 °С. В первый момент молекул водорода и кислорода ещё не имеется и u1 = 0. Наоборот, скорость u2 велика, так как молекул воды много. В следующий момент, когда часть их успела разложиться, скорость u1 становится уже заметной, а скорость u2 несколько уменьшается. По мере дальнейшего разложения воды u1 продолжает увеличиваться, u2 — уменьшаться. Наконец, наступает такой момент, когда обе скорости становятся равными.
Если исходить не из водяного пара, а из водорода и кислорода, то подобным же образом приходим к тем же результатам. И в том и в другом случае при равенстве скоростей обеих реакций устанавливается химическое равновесие, внешне характеризующееся тем, что концентрации водорода, кислорода и водяного пара при неизменных условиях остаются постоянными сколь угодно долгое время.
Из рассмотренного вытекает, чтохимическое равновесие является равновесием динамическим; оно обусловлено не тем, что, дойдя до него, процесс прекращается, а тем, что обе взаимно противоположные реакции протекают с одинаковыми скоростями. Всё время идёт и образование молекул воды, и их распад, но число образующихся за единицу времени молекул равно числу распадающихся. Поэтому нам и кажется, что изменений в системе не происходит.
Для изучения химических равновесий применяется ряд различных методов. Одним из наиболее общих является “замораживание” равновесий. Метод основан на том, что при достаточно низких температурах скорость реакций падает практически до нуля. Если, например, в тугоплавкой металлической трубке поместить смесь водорода с кислородом и выдержать её некоторое время при 2500 °С, то установится соответствующее этой температуре равновесие между исходными газами и водяным паром. При очень быстром охлаждении трубки равновесие не успевает сместиться, а в дальнейшем оно не смещается из-за крайне малой скорости реакции при низких температурах. Благодаря этому анализ содержимого трубки даст результаты, соответствующие положению равновесия при 2500 °С. Для контроля опыт повторяют, достигая равновесия с другой стороны — в нашем примере вводя первоначально в трубку не смесь водорода с кислородом, а воду. Результаты обоих опытов должны совпасть, так как одно и то же положение равновесия одинаково достижимо с обеих сторон.
Пользуясь выведенными выше выражениями для скоростей прямой и обратной реакций, можно подойти к важному понятию о константе равновесия. Так, при равновесии u1 = u2, откуда имеем
k1[H2]2[O2] = k2[H2O]2.
Для разъединения концентраций и констант скоростей делим обе части равенства на k2[H2]2[O2] и получаем:
k1/k2 = [H2O]2/[H2]2[O2].
Но частное от деления двух постоянных (при данных внешних условиях) величин — k1 и k2 — есть также величина постоянная. Она называется константой равновесия и обозначается буквой К. Таким образом:
[H2O]2/ [H2]2[O2] = К
Из изложенного вытекает практическое правило для составления выражений констант равновесия: в числителе дроби пишется произведение концентраций веществ правой части уравнения реакции, в знаменателе — левой части (или наоборот). При этом концентрация каждого вещества вводится в степени, равной числу его частиц, входящих в уравнение реакции. Числовое значение константы характеризует положение равновесия при данной температуре и не меняется с изменением концентраций реагирующих веществ.
В качестве простейшего примера экспериментального определения константы равновесия могут быть приведены полученные при 445 °С данные для реакции:
H2 + I2Ы 2 HI
При подходе с обеих сторон в рассматриваемой системе приблизительно через 2 ч достигается одно и то же равновесное состояние (78 % HI, 11 % H2 и 11 % паров I2 по объёму). Найденные на опыте значения равновесных концентраций (в моль/л) при различных неэквивалентных соотношениях реагирующих веществ и вычисленные из них величины константы равновесия сопоставлены ниже:
[H2] | 0,0268 | 0,0099 | 0,0032 | 0,0017 | 0,0003 | |
[H2][I2]/[HI]2 = K | [I2] | 0,0002 | 0,0020 | 0,0078 | 0,0114 | 0,0242 |
| [HI] | 0,0177 | 0,0328 | 0,0337 | 0,0315 | 0,0202 | |
| K | 0,017 | 0,018 | 0,022 | 0,020 | 0,018 |
Как видно из приведённых данных, несмотря на значительные колебания относительных концентраций H2 и I2, постоянство величины К сохраняется довольно хорошо. Среднее её значение может быть принято равным 0,02.
Связанные с константами равновесий количественные расчёты составляют предмет одного из важнейших отделов физической химии. Но даже в качественной форме выражение для константы равновесия даёт ценные указания по вопросу о взаимном влиянии концентраций отдельных компонентов равновесной системы.
Пусть в систему 2 Н2 + О2Ы 2 Н2О вводится избыток водорода. Постоянство значения константы равновесия может быть при этом сохранено только в том случае, если соответственно уменьшится концентрация кислорода и увеличится концентрация водяного пара. Практически это означает, что, желая при данных внешних условиях полнее использовать кислород, следует увеличивать концентрацию водорода. С другой стороны, чтобы полнее использовать водород, нужно вводить в систему избыток кислорода.
Того же эффекта — лучшего использования одного из реагирующих веществ — можно иногда добиться и путём уменьшения концентрации другого участника реакции. Допустим, что система 2 Н2 + О2Ы 2 Н2О заключена в сосуде, непроницаемом для водяного пара и кислорода, но пропускающем водород. Тогда последний будет покидать систему, уменьшая тем самым знаменатель выражения для константы равновесия. В силу постоянства К неизбежным результатом этого явится дальнейшее разложение водяного пара и накопление в сосуде свободного кислорода.
До сих пор равновесные системы рассматривались при неизменных внешних условиях. Общую формулировку влияния их изменения даёт принцип смещения равновесий (Ле-Шателье, 1884 г.), который может быть выражен следующим образом: если на равновесную систему производить внешнее воздействие, то равновесие смещается в сторону, указываемую этим воздействием, и до тех пор, пока нарастающее в системе противодействие не станет равно внешнему действию.
Общая формулировка принципа смещения равновесий наглядно иллюстрируется на примере следующей механической системы. Представим себе пружину, вделанную в неподвижную опору. Предоставленная самой себе, подобная система находится в равновесии. Если прилагать какую-то определённую внешнюю силу для растяжения пружины, то равновесие системы смещается в сторону, указываемую внешним воздействием, — пружина растягивается. Однако при этом возникают и по мере деформации пружины всё более увеличиваются силы её упругости, т. е. в системе нарастает противодействие. Наконец, наступает такой момент, когда это противодействие становится равным внешнему действию: устанавливается новое равновесное состояние, отвечающее растянутой пружине, т. е. смещение относительно исходного в сторону, указываемую внешним воздействием.
Принцип смещения равновесий необычайно широк. Именно поэтому его общая формулировка несколько расплывчата. Ниже этот принцип детально рассматривается в применении к важнейшим для химии внешним условиям — температуре и давлению.
Уравнение
2 Н2 + О2 = 2 Н2О + 485 кДж
показывает, что соединение водорода с кислородом сопровождается выделением тепла, а распад водяного пара на элементы — его поглощением. Если мы имеем рассматриваемую систему в равновесии при некоторой температуре и затем нагреваем её, то равновесие последовательно смещается в сторону образования всё больших концентраций свободного водорода и кислорода. Но по закону действия масс одновременно ускоряется и идущая с выделением тепла реакция их соединения, т. е. в системе постоянно нарастает противодействие. Новое равновесие устанавливается тогда, когда концентрации свободных водорода и кислорода возрастут настолько, что выделяемое при их взаимодействии количество тепла станет равно сообщаемому за то же время системе извне.
Чем больше тепла сообщается системе, тем более это благоприятствует распаду водяного пара, т. е. эндотермической реакции. Наоборот, отвод тепла от системы благоприятствует более полному соединению водорода с кислородом, т. е. экзотермической реакции. Следовательно, при нагревании равновесной системы равновесие смещается в сторону эндотермической реакции, при охлаждении — в сторону экзотермической.
Для газообразной системы
2 Н2 + О2Ы 2 Н2О
имеем в левой части уравнения 3 молекулы, в правой — 2 молекулы. Применяя закон Авогадро, находим, что если бы весь водяной пар разложился на водород и кислород, то система занимала бы 3 объёма, а если бы распада совсем не было — 2 объёма. Фактически занимаемое системой число объёмов должно быть некоторым промежуточным, зависящим от положения равновесия, причём смещение последнего в сторону образования водяного пара ведёт к уменьшению объёма, а в сторону его распада — к увеличению.
Изменение оказываемого на газообразную систему внешнего давления должно вызывать соответствующее изменение её объёма. При повышении давления он будет уменьшаться, при понижении — увеличиваться. Допустим, что оказываемое на систему давление повышается. Равновесие при этом смещается в сторону образования водяного пара, т. е. его относительная концентрация возрастает. Но по закону действия масс соответственно ускоряется идущее с увеличением объёма разложение водяного пара на элементы. Результатом этого является нарастание в системе противодействия. Новое состояние равновесия установится при такой концентрации водяного пара, когда создаваемое самой системой давление станет равно производимому на неё извне.
Таким образом, при увеличении внешнего давления на систему 2 Н2 + О2Ы 2 Н2О равновесие сместится в сторону образования воды, при уменьшении — в сторону её распада. Сопоставляя данные при какой-либо одной температуре, можно видеть влияние на диссоциацию увеличения и уменьшения давления.
Подобно рассмотренному выше случаю диссоциации воды, внешнее давление влияет и на положение равновесия других обратимых реакций между газами, протекающих с изменением объёма. Последнее же обусловлено разным числом молекул в левой и правой частях уравнения реакции.
Отсюда вытекает формулировка принципа смещения равновесий применительно к влиянию давления на равновесие обратимых газовых реакций: при увеличении давления равновесие смещается в сторону образования меньшего числа молекул, при уменьшении — в сторону большего. Если общее число молекул в левой и правой частях уравнения реакций одинаково, изменение давления не влияет на положение химического равновесия.
Независимость химического равновесия от давления в газообразных системах с неизменным числом молекул вполне верна для идеальных газов. Так как реальные газы обладают несколько различной сжимаемостью, на самом деле равновесие таких систем зависит от давления. Однако эта зависимость становится заметной лишь при высоких давлениях.
Так как занимаемые твёрдыми и жидкими веществами объёмы лишь очень мало меняются в процессе реакции, изменение давления почти не влияет на равновесия подобных (“конденсированных”) систем. В смешанных случаях, когда одновременно имеются вещества различных агрегатных состояний, для учёта влияния давления на равновесие практическое значение обычно имеет только число молекул газообразных веществ.
Пример. Пусть имеется система СО2 + С Ы 2 СО. Подходя к подсчёту числа частиц формально (2 слева и 2 справа), можно было бы сделать вывод, что давление не влияет на равновесие данной системы. Однако газами являются только СО2 и СО (С — твёрдое вещество). Поэтому повышение давления будет смещать рассматриваемое равновесие влево, а понижение давления — вправо.
Если равновесие какой-либо обратимой реакции очень сильно смещено в одну сторону, то она представляется нам при данных условиях необратимой, т. е. способной протекать только в одном направлении. Например, когда водород соединяется с кислородом при 1000 °С, нам кажется, что свободных молекул водорода и кислорода совершенно не остаётся. На самом деле ничтожно малое количество этих молекул всё же имеется, причём за единицу времени их столько же образуется из водяного пара, сколько соединяется.
Таким образом, в действительности обратимые реакции являются таковыми во всём интервале температур, при которых вообще могут существовать рассматриваемые вещества. Практически же обратимость заметна лишь в некотором более узком интервале, например для реакции образования воды (под обычным давлением) между 2000 и 4000 °С.
При таком более широком рассмотрении подавляющее большинство химических реакций оказывается принадлежащим к типу обратимых, но часто с настолько смещённым в одну сторону равновесием, что их обратимость практически незаметна до тех пор, пока соответственно не изменяются внешние условия. Именно эта распространённость обратимых реакций обусловливает особую важность для химии изучения равновесий и условий их смещения.
Помимо температуры и давления, на химическое равновесие могут в большей или меньшей степени влиять и различные другие факторы. Например, было показано, что равновесие газообразной системы 2 Н2 +О2Ы 2 Н2О несколько смещается вправо под действием электрического поля. Обусловлено это полярным характером молекул воды при неполярности молекул водорода и кислорода.
Чем значительнее мы изменим какой-либо влияющий на равновесие фактор, тем сильнее сместится химическое равновесие. Вместе с тем при изменении этого фактора на очень малую величину сместится и равновесие. Таким образом, процесс его смещения представляется нам непрерывным. Однако эта непрерывность лишь кажущаяся: она обусловлена тем, что даже самое незначительное замечаемое нами химическое изменение связано с превращением миллиардов молекул. Очевидно, что не может произойти смещения равновесия меньшего, чем даёт химическое изменение одной молекулы, а всякое иное должно соответствовать превращению целого числа молекул. Следовательно, в действительности смещение равновесия идёт скачками, но настолько малыми, что для нас ряд таких скачков сливается в один непрерывный процесс.
Шестая группа периодической системы
Атомы элементов VI группы характеризуются двумя различными структурами внешнего электронного слоя содержащего либо шесть, либо одного или двух электронов. К первому типу, помимо кислорода, относится сера и элементы подгруппы селена (Se, Te, Po), ко второму — элементы подгруппы хрома (Cr, Mo, W).
Структура внешнего слоя атомов серы, селена и его аналогов придает им преимущественно неметаллический характер с максимальной отрицательной валентностью, равной двум. Эти элементы должны быть менее активными неметаллами, чем галогены (так как последним не хватает до устойчивой конфигурации лишь по одному электрону). Максимальную положительную валентность серы, селена и его аналогов можно ожидать равной шести, причём электроны должны отдаваться ими легче, чем стоящими в том же горизонтальном ряду галогенами.
Наличие во внешнем слое атомов лишь одного или двух электронов обуславливает металлический характер элементов подгруппы хрома. Вместе с тем их максимальная положительная валентность также должна быть равна шести.
Сера
Элемент этот был известен ещё древним египтянам. В теоретических представлениях алхимиков сера играла большую роль, так как считалась наиболее совершенным выразителем одного из “основных начал” природы — горючести.
По содержанию в земной коре (0,03 %) она относится к весьма распространённым элементам. Формы нахождения серы в природе многообразны. Сравнительно редко встречаются её самородные месторождения, основная же масса серы связана с металлами в составе различных минералов, которые могут быть разбиты на две большие группы: сульфидов и сульфатов. Из сульфидов особое значение для технологии серы имеет пирит FeS2. Из сульфатов наиболее распространен гипс CaSO4·2H2O. Соединения серы обычно присутствуют в вулканических газах и воде некоторых минеральных источников. Сера входит также в состав белковых веществ и поэтому содержится в организмах животных и растений.
Сера метеоритного происхождения состоит из четырёх изотопов: 32 (95,0 %), 33 (0,76 %), 34 (4,22 %), 36 (0,02 %). Изотопный состав серы различных земных объектов очень близок к приведённому, но не вполне постоянен. В этом отношении сера подобна кислороду, состав которого в воздухе (99,76 % 16О; 0,04 %17О; 0,20 %18О) и других природных объектах обнаруживает ничтожные, но все же заметные расхождения.
В основном состоянии атом серы имеет структуру внешнего электронного слоя 3s23p4 и, подобно кислороду, двухвалентен. Возбуждение четырёхвалентного состояния (3s23p34s1) требует затраты 627 кДж/моль, что приблизительно на 251 кДж/моль меньше, чем у кислорода.
Растения накапливают серу главным образом в семенах и листьях. Например, капуста содержит около 0,8 % серы (по расчету на сухое вещество). У животных особенно велико её содержание в волосах (до 5 %), костях, рогах и копытах. Интересно, что состав золы волос существенно зависит от их цвета.
Свободная сера может быть получена либо из её самородных месторождений, либо из соединений. Почти вся мировая выработка осуществляется по первому варианту, причём технологический процесс сводится к отделению серы от смешанных с нею пород (песка, глины и т. п.), что может быть проще всего достигнуто выправлением серы.
В древности и в средние века серу добывали из её самородных месторождений весьма примитивным способом. В землю вкапывали глиняный горшок, на него ставили другой горшок с отверстиями в дне. Последний заполняли содержащей серу породой и затем нагревали снаружи. При этом сера плавилась и стекала в нижний горшок.
В настоящее время выплавка самородной серы производится обычно обработкой исходной (или предварительно обогащённой) руды нагретым до 140-150 °С водяным паром. Реже применяется нагревание руды за счёт сжигания части содержащейся в ней серы. Много серы получают в настоящее время из металлургических и нефтяных газов, а также при очистке нефти от примеси сернистых соединений.
Некоторые очень богатые месторождения серы долгое время не находили промышленного использования из-за особых условий их залегания — под толстыми слоями песка, на глубине 200-300 м. Этот песок и выделяющийся из сероносных пластов сероводород не давали возможности проложить шахты и вести работу в них.
Рис. 1. Схема установки для подземной выплавки серы.
Положение изменилось лишь в начале текущего столетия, с изобретением способ выплавки серы под землей и извлечения её на поверхность в жидком состоянии. Способ этот основан на легкоплавкости серы и её сравнительно небольшой плотности. В серный слой вводятся три трубы одна в другой (рис. 1). По внешней трубе пропускается вода нагретая до 170 °С (под давлением). Попадая в руду она расплавляет серу, которая собирается в образующемся под трубами углублении. Нагнетаемый по внутренней трубе горячий воздух вспенивает жидкую серу и по средней трубе гонит её на поверхность, где она вытекает в огороженное досками пространство, постепенно образуя громадные массивы.Метод подземной выплавки применим только к достаточно мощным и богатым месторождениям. Требуя большого расхода воды и топлива, он вместе с тем позволяет извлекать лишь около 50 % всей имеющейся в руде серы.
Получаемая из природных месторождений сера обычно содержит примеси. Для очистки её подвергают перегонке в специальных печах или перекристаллизовывают из сероуглерода. Сера, очищенная перекристаллизацией, часто содержит примесь встроенного в кристаллы растворителя. Глубокая очистка серы может быть достигнута многократно повторённым длительным нагреванием её расплава в присутствии оксида магния. Очень чистая сера совершенно не имеет запаха и весьма склонна к переохлаждению.
Ежегодное мировое потребление серы составляет около 20 млн. т. Её промышленными потребителями являются самые разнообразные производства: сернокислотное, бумажное, резиновое, спичечное и др. Сера широко используется для борьбы с вредителями сельского хозяйства, в пиротехнике и отчасти в медицине.
Чистая сера представляет собой жёлтое кристаллическое вещество с плотностью 2,1 г/см3, плавящееся при 119 °С и кипящее при 445 °С. Она очень плохо проводит тепло и электричество. В воде сера нерастворима. Лучшим её растворителем является сероуглерод (СS2).
Элементарная сера существует при обычных условиях в виде восьмиатомных кольцевых молекул (рис. 2). Кристаллы образованные молекулами S8 имеют две формы. Ниже 95,4 °С устойчива обычная жёлтая сера с плотностью 2,07 г/см3, кристаллизующаяся в ромбической системе и имеющая т. пл. 112,8 °С (при быстром нагревании). Напротив, выше 95,4 °С устойчивы бледно-жёлтые кристаллы моноклинной серы с плотностью 1,96 г/см3 и т. пл. 119,3 °С (Sb) (рис. 3). Теплота превращения одной формы в другую составляет 3 кДж/моль. Интересно, что при трении сера приобретает сильный отрицательный заряд, а при охлаждении ниже -50 °С обесцвечивается.
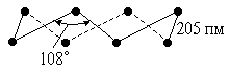
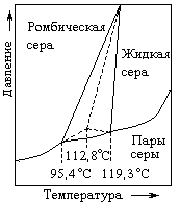
Рис. 2. Строение молекулы S8. Рис. 3. Диаграмма состояния серы.
В особых условиях удавалось получать для серы малоустойчивые разновидности и иных типов. Например, при замораживании (жидким азотом) сильно нагретых паров серы получается ее устойчивая лишь ниже -80 °С пурпурная модификация, образованная молекулами S2. Лучше других изучена форма, извлекаемая толуолом из подкисленного раствора Nа2S2O3. Ее оранжево-жёлтые кристаллы образованы кольцеобразными молекулами S6 [с параметрами d(SS) = 206 пм и РSSS = 102°]. Резким охлаждением насыщенного раствора серы в бензоле может быть получена состоящая из молекул S8 метастабильная ”перламутровая” модификация (Sg). Довольно сложным путем была получена форма, слагающаяся из циклических молекул S12 [d(SS) = 206 пм, РSSS = 106,5°]. Известны также формы, образованные молекулами S7 и S10.
Имеющиеся пока сведения о поведении серы при высоких давлениях неполны и отчасти противоречивы. Так, по одним данным, в области около 30 тыс. атм. и 300 °С формы кристаллов серы. устойчива кубическая фаза с плотностью 2,18 г/см3, по другим — в той же области устойчива обладающая ромбической решеткой “волокнистая” сера, слагающаяся из десятиатомных спиралей (с тремя оборотами на период 138 пм). Предполагается, что при 200 тыс. атм. сера может быть переведена в металлическое состояние.
Теплота плавления серы составляет 1,3 кДж/моль. Плавление сопровождается заметным увеличением объема (примерно на 15 %). Расплавленная сера представляет собой желтую легкоподвижную жидкость, которая выше 160 °С превращается в очень вязкую темно-коричневую массу. Как видно из рис. 4, около 190 °С вязкость серы примерно в 9000 раз больше, чем при 160 °С. Затем она начинает уменьшаться, и выше 300 °С расплавленная сера, оставаясь темно-коричневой, вновь становится легко подвижной.
Изменений физических свойств при нагревании связано с изменением внутреннего строения серы. Выше 160 °С кольца S8 начинают разрываться, причем концевые атомы возникающих открытых структур сцепляются друг с другом, образуя цепи с длиной до миллиона атомов, что сопровождается резким повышением вязкости (и изменением цвета). Дальнейшее нагревание ведет к быстрому уменьшению средней длины цепей, вследствие чего вязкость уменьшается (хотя все же остается значительно большей, чем ниже 160 °С). Работа разрыва цепи оценивается в 138 кДж/моль.
Температура кипения серы (444,7 °С) является одной из вторичных стандартных точек международной шкалы. Теплота испарения серы составляет 9,2 кДж/моль. В парах имеет место равновесие главным образом между молекулами S8, S6, S4 и S2, причем переход от S8 к S2 осуществляется эндотермически:
1/4S8 (+4,4 кДж) ® 1/3S6 (+29 кДж) ®1/2S4 (+58 кДж) ® S2
Поэтому по мере повышения температуры равновесие все более смещается вправо. Внешним признаком этого служит изменение цвета паров, которые вблизи точки кипения имеют оранжево-желтую окраску, при дальнейшем нагревании сначала краснеют, а затем начинают бледнеть и при 650 °С становятся соломенно-желтыми. Пары кипящей серы содержат (по объему) приблизительно 59 % S8, 34 % S6, 4 % S4 и 3 % S2 с небольшими примесями нечетных молекул (S3, S5, S7). По видимому, все эти молекулы Sn (кроме S2) имеют циклическое строение. Около 900 °С пары серы состоят практически только из молекул S2 [с расстоянием d(SS) = 189 пм]. Энергия их диссоциации на атомы равна 418 кДж/моль и заметной она становится приблизительно с 1500 °С. Теплота атомизации серы (при 25 °С) равна 272 кДж/моль.
По электронному строению молекула S2 подобна молекуле О2. Магнитные свойства последней указывают на наличие в ней двух неспаренных электронов.
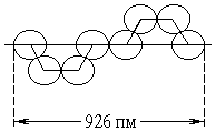
Рис. 5. Вероятное строение цепи Sm.
При быстром охлаждении жидкой серы или ее пара существующие в них равновесия не успевают сместиться. Образующаяся твердая фаза (т. пл. около 110 °С) содержит серу двух типов — растворимую (Sl) и нерастворимую (Sm) в сероуглероде. Примесью Smобусловлена, в частности, неполная растворимость в СS2, серного цвета. Еще гораздо больше Smсодержит похожая по тягучести на резину коричневая ”пластическая” сера, получаемая вливанием ее нагретого выше 300 °С расплава в холодную воду. Вытянутые нити пластической серы слагаются из цепей Sm строение которых показано на рис. 5. При хранении пластическая сера быстро твердеет, но полностью переходит в Sl крайне медленно.Данные по растворимости серы (Sa) в сероуглероде и бензоле приводятся ниже (г серы в 100 г насыщенного раствора):
Температура, °С | 20 | 40 | 60 | 80 | 100 | |
Растворимость в СS2 | 18,0 | 29,5 | 50,0 | 66,0 | 79,0 | 92,0 |
Растворимость в С6Н6 | 1,0 | 1,7 | 3,2 | 6,0 | 10,5 | 17,5 |
Хорошо растворяется сера в скипидаре. Более или менее растворима она и во многих других органических жидкостях. Например, 100 г эфира растворяют при обычных условиях около 0,2 г серы.
Чистая сера не ядовита. Прием внутрь небольших ее количеств способствует рассасыванию нарывов и полезен, в частности, при геморрое. В дозах порядка 1 г она иногда назначается как слабительное. Организм человека не обнаруживает привыкания к сере, но длительное ее потребление может неблагоприятно отразиться на работе печени и кишечника. Очень мелко раздробленная (осажденная) сера входит в состав ряда мазей, предназначаемых для ухода за кожей и лечения кожных заболеваний.
Интересны опыты использования серы в строительстве. Расплавленную серу смешивают со стеклянным волокном и охлаждают. Получается прочный строительный материал, не пропускающий влагу и холод.
Сера может служить простейшим примером электрета, вещества, способного длительно сохранять электрический заряд (в том числе разного знака на противоположных поверхностях) и создавать электрическое поле в окружающем пространстве. Электретное состояние обычно достигается нагреванием и последующим охлаждением пластин из подходящего вещества в достаточно сильном электрическом поле. Электреты являются как бы электрическими аналогами постоянных магнитов и находят разнообразное практическое использование.
Наиболее характерным для серы валентным состояниям отвечают значности -2, 0, +4 и +6. Схема окислительно-восстановительных потенциалов, соответствующих переходам между ними, дается ниже:
Значность -2 0 +4 +6
Кислая среда +0,14 +0,45 +0,17
Щелочная среда -0,48 -0,61 -0,91
На холоду сера сравнительно инертна (энергична соединяясь только со фтором), но при нагревании становится весьма химически активной — реагирует с хлором и бромом (но не с иодом), кислородом, водородом и металлами. В результате реакций последнего типа образуются соответствующие сернистые соединения, например:
Fe + S = FeS + 96 кДж
С водородом сера в обычных условиях не соединяется. Лишь при нагревании протекает обратимая реакция:
Н2 + S = H2S + 21 кДж
равновесие которой около 350 °С смещено вправо, а при повышении температуры смещается влево. Практически сероводород получают обычно действием разбавленных кислот на сульфид железа:
FeS + 2 HСl = FeCl2 + H2S
Молекула Н2S имеет структуру равнобедренного треугольника с атомом серы в центре [d(HS) = 133 пм, РHSH = 92°]. Сероводород представляет собой бесцветный и весьма ядовитый газ, уже 1 часть которого на 100 000 частей воздуха обнаруживается по его характерному запаху (тухлых яиц).
Один объём воды растворяет в обычных условиях около 3 объемов сероводорода (с образованием приблизительно 0,1 М раствора (сероводородной воды). При нагревании растворимость понижается. Подожженный на воздухе сероводород сгорает по одному из следующих уравнений:
2 H2S + 3 O2 = 2 H2O + 2 SO2 + 1125 кДж (при избытке кислорода)
2 H2S + O2 = 2 H2O + 2 S + 531 кДж (при недостатке кислорода).
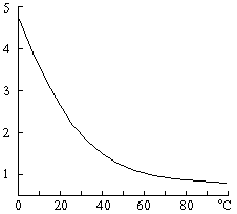
Рис. 6. Растворимость сероводорода (объемы на 1 объём воды).
Легко окисляется сероводород и в растворе: при стоянии на воздухе сероводородная вода постепенно мутнеет вследствие выделения серы (по второй из приведенных выше реакций). Бром и иод восстанавливаются до НВr и HI. Аналогично действует он и на многие другие вещества. Сероводород является, таким образом, сильным восстановителем.В водном растворе H2S ведёт себя как весьма слабая кислота. Средние соли (с анионом S2-) называются сульфидами, кислые (с анионом HS-) — гидросульфидами. Несмотря на бесцветность самих ионов S2- и HS-, многие соли сероводорода окрашены в характерные цвета. Подавляющее большинство сульфидов практически нерастворимо в воде. А большая часть гидросульфидов хорошо растворима (но известна лишь в растворе).
Удобный способ получения H2S состоит в нагревании выше 170 °С сплава порошкообразной серы с парафином и измельченным асбестом (приблизительно 3: 5: 2 по массе). При охлаждении реакция прекращается, но вновь вызывается нагреванием. Исходный сплав может заготовляться впрок и расходоваться по мере надобности (один грамм дает около 150 мл H2S).
Очень чистый сероводород (т. пл. -86, т. кип. -60 °С) может быть получен пропусканием смеси Н2 с парами серы над нагретыми до 600 °С кусками пемзы. Критическая температура H2S равна 100 °С при критическом давлении 89 атм. Термическая диссоциация H2S начинается приблизительно с 400 °С и становится практически полной около 1700 °С.
Сульфгидрильные группы (HS) входят в состав некоторых биологически важных органических соединений.
В жидком состоянии H2S проводит электрический ток несравненно хуже, чем вода, так как собственная его электролитическая диссоциация ничтожно мала: [SH3+] [HS-] = 3·10-33. Жидкий, сероводород имеет низкую диэлектрическую проницаемость (e = 6 при 0 °С) и как растворитель похож скорее на органические жидкости, чем на воду. Так, он практически не растворяет лед. Твердый Н2S имеет строение плотной упаковки с 12 ближайшими соседями у каждой молекулы (т. е. совершенно иное, чем лед). Теплота плавления сероводорода равна 2,5 кДж/моль, а теплота испарения 18,8 кДж/моль.
На воздухе сероводород воспламеняется около 300 °С. Взрывоопасны его смеси с воздухом, содержащие от 4 до 45 объемн. % H2S. Ядовитость сероводорода часто недооценивают и работы с ним ведут без соблюдения достаточных мер предосторожности. Между тем уже 0,1 % H2S в воздухе быстро вызывает тяжелое отравление. При вдыхании сероводорода в значительных концентрациях может мгновенно наступить обморочное состояние или даже смерть от паралича дыхания (если пострадавший не был своевременно вынесен из отравленной атмосферы). Первым симптомом острого отравления служит потеря обоняния. В дальнейшем появляются головная боль, головокружение и тошнота. Иногда через некоторое время наступают внезапные обмороки. Противоядием служит прежде всего чистый воздух. Тяжело отравленным сероводородом дают вдыхать кислород. Иногда приходится применять искусственное дыхание. Хроническое отравление малыми количествами H2S обусловливает общее ухудшение самочувствия, исхудание, появление головных болей и т. д. Предельно допустимой концентрацией H2S в воздухе производственных помещений считается 0,01 мг/л. Содержащие его баллоны должны иметь белую окраску с красной надписью “Сероводород” и красной чертой под ней.
Охлаждение насыщенного водного раствора сероводорода может быть получен кристаллогидрат Н2S·6Н2О. Растворимость Н2S в органических растворителях значительно выше, чем в воде. Например, один объем спирта поглощает при обычной температуре 7 объемов сероводорода. Растворимость его в расплавленной сере резко возрастает выше 130 °С и достигает максимума около 350 °С. По-видимому, это связано с образованием полисульфидов.
В водном растворе сероводород легко окисляется иодом до свободной серы: I2 + Н2S = 2 НI + S. Напротив, в газовой фазе сера окисляет иодистый водород до свободного иода: S +2 НI = Н2S + I2 + 6 кДж. Ниже -50 °С может существовать молекулярное соединение состава Н2S·I2.
Сероводородная кислота характеризуется константами диссоциации К1 = 1·10-7 и К2 = 1·10-14, т. е. она несколько слабее угольной. Децинормальный раствор H2S имеет рН = 4,1.
Помимо прямого соединения металла с серой и реакции нейтрализации, многие сульфиды (малорастворимые) могут быть получены обменным разложением в растворе солей соответствующего металла с H2S или (NН4)2S. Часто применяемый в лабораториях раствор последней соли готовят обычно, насыщая сероводородом раствор NН4OН (что дает NН4SН) и смешивая затем полученную жидкость с равным объемом NH4ОН.
Раствор (NН4)2S характеризуется значением рН около 9,3. В нем имеет место гидролитическое равновесие: S” + НОН Ы SН’+ ОН’. Так как оно смещено вправо, раствор содержит очень мало ионов S”. Поэтому образование малорастворимых сернистых металлов при обменном разложении их солей с сернистым аммонием идет в основном по схеме (для двухвалентного катиона): М2+ + SН-Ы МSН+Ы МS + Н+. Очевидно, что равновесие должно смещаться вправо тем сильнее, чем менее растворим сульфид и меньше концентрация водородных ионов.
На различной растворимости в воде сернистых соединений отдельных металлов основан систематический ход качественного анализа катионов. Часть последних (Nа+, К+, Вa2+ и т. д.) образует сульфиды, растворимые в воде, другая часть (Fе2+, Мn2+, Zn2+ и т. д.) — нерастворимые в воде, но хорошо растворимые в разбавленной НСl, и, наконец, третья часть (Сu2+, Рb2+, Нg2+ и т. д.) — нерастворимые ни в воде, ни в разбавленных кислотах. Поэтому, действуя на раствор смеси катионов сероводородом сначала в кислой среде, затем в нейтральной или слабощелочной, можно отделить группы катионов друг от друга и дальше вести анализ уже в пределах каждой из них отдельно.
По отношению к нагреванию в отсутствие воздуха большинство сульфидов весьма устойчиво. Прокаливание их на воздухе сопровождается переходом сульфида в оксид или сульфат. При температурах порядка 1200 °С многие сульфиды восстанавливаются до металла углем (по схеме 2 ЭS + С = СS2 + 2 Э).
При внесении в крепкий раствор сульфида мелко растертой серы она растворяется с образованием соответствующего полисульфида (многосернистого соединения), например: (NН4)2S + (n-1)S = (NН4)2Sn. Обычно образуется смесь полисульфидов с различным содержанием серы. По мере увеличения n цвет соединения меняется от желтого через оранжевый к красному. Интенсивно красную окраску имеет и самое богатое серой из соединений этого типа — (NН4)2S9.
Полисульфиды аммония применяются для воронения стали. Смесью полисульфидов натрия в виде “серной печени” пользуются в кожевенной промышленности для снятия волоса со шкур. Серную печень для этой цели готовят сплавлением серы с содой. Получающаяся зеленовато-коричневая масса растворяется в воде с сильнощелочной реакцией и при стоянии раствора постепенно разлагается с выделением сероводорода. Некоторые органические производные полисульфидного типа находят применение в качестве горючего твердых реактивных топлив.
Некоторые полисульфиды хорошо кристаллизуются (часто с кристаллизационной водой). В твердом состоянии они очень гигроскопичны и во влажном воздухе легко окисляются. Образование полисульфидов имеет место также при медленном окислении растворов сернистых солей кислородом воздуха, например, по схеме:
4 НS- + О2 = 2 Н2О + 2 S22-
Для предохранения растворов (NН4)2S от образования полисульфидов рекомендуется добавлять к ним немного Zn или Сu.
Если крепкий раствор полисульфида небольшими порциями вылить в избыток раствора НСl, на дне сосуда собирается тяжелое желтое масло, представляющее собой смесь сульфанов (многосернистых водородов) общей формулы Н2Sn. Они более или менее устойчивы лишь в сильнокислой среде, а при других условиях разлагаются с выделением серы. Существовать могут, по-видимому, сульфаны с очень большими значениями n. В индивидуальном состоянии получены все члены ряда вплоть до Н2S8. Они являются маслянистыми желтыми жидкостями с резким запахом. Лучше других охарактеризованы Н2S2 (т. пл. -88, т. кип. 75 °С), Н2S3 (т. пл. -52 °С), Н2S4 (т. пл. -85 °С) и Н2S5 (т. пл. -50 °С).
Молекулы многосернистых водородов образованы зигзагообразными цепями из атомов серы с атомами водорода на концах. По-видимому, такие цепи никогда не бывают разветвленными (отличие от углерода). Энергии связей S-S (263 кДж/моль) и S-Н (347 кДж/моль) довольно постоянны для всех сульфанов. Последние являются кислотами и тем более сильными, чем больше в них атомов серы. Это доказывается уменьшением в том же направлении степени гидролиза их солей, как видно из приводимых ниже данных для Nа2Sn (в 0,1 н. растворах при 25 °С):
n 1 2 3 4 5
Степень гидролиза, % 86,4 64,6 37,6 11,8 5,7
Значения констант диссоциации определены для Н2S4 (К1=2·10-4, К2 = 5·10-7) и Н2S5 (К1 = 1·10-4, К2 = 3·10-6). Водой и спиртом сульфаны более или менее легко разлагаются, а в эфире и бензоле растворяются без разложения.
Дисульфан по строению подобен пероксиду водорода. Его молекула характеризуется следующими параметрами: d(НS) = 135, d(SS) = 206 пм, РНSS = 92° при угле 91° между связями Н-S. Барьер свободного вращения по связи S-S равен 12,6 кДж/моль. Жидкий двусернистый водород хорошо растворяет серу, причем растворение не сопровождается образованием высших сульфанов. Из природных многосернистых соединений наиболее известен минерал пирит (FеS2), представляющий собой железную соль двусернистого водорода. Подобную же структуру имеет и МnS2.
Взаимодействие Н2S с бромом в хлороформе может быть проведено по схеме Вr2 + Н2S Ы НВr + НSВr с образованием бромистого водорода и тиобромноватистой кислоты. Введение в систему сухого аммиака смещает равновесие вправо, так как получаются NH4Вr и NН4SВr. Последняя соль устойчива лишь при низких температурах, а в обычных условиях постепенно разлагается на NН4Вr и S. Аналогично брому ведет себя и иод. Это особенно интересно потому, что непосредственно с серой иод не соединяется.
Сродство серы к галогенам в ряду F-Cl-Br-I настолько быстро уменьшается, что ее иодистое производное получить вообще не удается. С остальными галогенидами она соединяется более или менее легко.
Некоторые свойства галогенидов серы сопоставлены ниже:
SF6 | S2F10 | SF5Cl | SF4 | S2F2 | SCl4 | SCl2 | S2Cl2 | S2Br2 | |
| Агрегатное состояние | газ | жидк. | газ | газ | газ | нест. | жидк. | жидк. | жидк. |
| Цвет | бесцв. | бесцв. | бесцв. | бесцв. | бесцв. | бесцв. | красн. | бесцв. | красн. |
Температура плавления °С | -50 давл. | -53 | -64 | -121 | -133 | -30 | -121 | -77 | -40 |
Температура кипения, °С | -64 возг. | +29 | -19 | -37 | +15 | (разл.) | +60 | +138 | +57 (27 Па) |
Большинство этих соединений образуется при взаимодействии элементов и легко разлагается водой.
Из образующихся соединений наиболее интересен газообразный при обычных условиях гексафторид SF6. Он бесцветен, не имеет запаха и не ядовит. От других галогенидов серы SF6 отличается своей исключительной химической инертностью. Как очень хороший газообразный изолятор, он находит применение в высоковольтных установках.
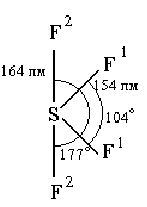
Рис. 7. Строение молекулы SF4.
Он образуется из элементов с большим выделением тепла (1208 кДж/моль) и термически устойчива до 800 °С. Его критическая температура равна 46 °С при критическом давлении 38 атм. Молекула SF6 неполярна и характеризуется высоким значением потенциала ионизации (19,3 в). Ее сродство к электрону оценивается в 142 кДж/моль. По строению она представляет собой октаэдр с серой в центре [d(SF) = 156 пм]. Средняя энергия связи S-F равна 322 кДж/моль.Будучи менее всех других газов растворима в воде (примерно 1:200 по объему), гексафторид серы не реагирует не только с ней, но и с растворами NаОН или НСl. Металлическим натрием он разлагается лишь при повышенных температурах (но быстро реагирует при -64 °С с его растворами в жидком аммиаке). С водородом и кислородом SF6 не взаимодействует, но при нагревании ее с Н2S до 400 °С идет реакция по схеме:
SF6 + 3 Н2S = 6 НF + 4 S
Иодистым водородом SF6 легко разлагается по схеме:
SF6 + 8 НI = 6 НF + Н2S + 4I2
уже при 30 °С.
Фторид S2F10 образуется при взаимодействии элементов (теплота образования 2132 кДж/моль) в качестве примеси к SF6. По химическим свойствам S2F10 в общем похож на SF6, но отличается меньшей инертностью и очень ядовит (более, чем фосген). Взаимодействием S2F10 с Сl2, при нагревании может быть получен фторохлорид SF5Cl [d(SF) = 158, d(SСl) = 203 пм]. Он медленно гидролизуется водой и быстро — щелочами.
Тетрафторид серы (теплота образования из элементов 769 кДж/моль, энергия связи SF 339 кДж/моль) может быть получен по проводимой при 200-300 °С под давлением реакции:
S + 2 Cl2 + 4 NaF = 4 NaCl + SF4
Его молекула полярна и имеет строение деформированной тригональной бипирамиды (рис. 7), одна из позиций основания которой как бы заполнена свободной электронной парой атома серы. В отличие от инертного SF6, тетрафторид серы способен образовывать продукты присоединения, является хорошим фторирующим агентом, легко разлагается под действием воды и сильно ядовит. Совместным нагреванием СsF и SF4 (под давлением) был получен СsSF5.
Дихлорид серы образуется при пропускании хлора в S2Сl2 по обратимой реакции:
S2Сl2 + Сl2Ы 2 SСl2.
В обычных условиях она медленно разлагается на хлористую серу и хлор. Молекула SСl2 имеет форму равнобедренного треугольника с атомом серы в вершине [d(SCl) = 200 пм, РClSCl = 103°).
Четырёххлористая сера может быть получена действием на S2Сl2 жидкого хлора. Соединение это устойчиво лишь в твердом состоянии, а при плавлении распадается на SCl2 и Сl2. Значительно устойчивее некоторые продукты присоединения SСl4, например SСl4·SbСl5 (возг. при 125 °С). Водой тетрахлорид серы разлагается с образованием SO2 и НСl.
Бромистая сера образуется из элементов (теплота образования 16,8 кДж/моль) лишь при нагревании (в запаянной трубке). Под действием воды она разлагается, в основном по уравнению:
S2Br2 + 2 H2O = 2 HBr + H2S2O2 и
H2S2O2 + S2Br2 = 2 HBr + SO2 + 3S
(промежуточно возникающая и тотчас же разлагающаяся тиосернистая кислота — Н2S2O2 — сама по себе и в виде солей неизвестна, но некоторые ее органические производные были получены). Аналогично протекает взаимодействие с водой и S2Cl2. Последний при хранении постепенно разлагается и сначала желтеет, а затем становится красно-бурой. Монобромид сера еще менее устойчив (а при нагревании разлагается уже выше 90 °С). Интересно, что элементарная сера хорошо растворима в S2Cl2 (около 1: 4 по массе при обычных условиях), но почти нерастворима в S2Br2.
Получить в индивидуальном состоянии какое-либо соединение серы с иодом не удается. При совместном нагревании обоих элементов происходит лишь понижение температуры плавления системы (вплоть до 65 °С при 80 % серы).
Строение молекул хлорида и бромида серы типа S2Г2 долго оставалось спорным, причем обсуждению подвергались формулы S=SГ2 и Г-S-S-Г. Результаты структурного анализа говорят в пользу второй трактовки: и S2Сl2, и S2Вr2 по строению подобны пероксиду водорода и имеют параметры d(SS) = 197, d(SСl) = 207 пм, РSSСl = 107° — для S2Сl2 и d(SS) = 198, d(SВr) = 224 пм, РSSBr = 105° — для S2Вr2. Угол между плоскостями S-S-Сl составляет 88°, а энергетический барьер свободного вращения равен 71 кДж/моль. Энергия связи S-Сl оценивается в 255 кДж/моль.
Монохлорид серы (S2Сl2) получают в больших масштабах прямым действием сухого хлора на избыток серы (теплота образования из элементов 59 кДж/моль). Он является хорошим растворителем многих химических соединений. Высокие значения ее криоскопической (5,36 град) и эбуллиоскопической (5,02 град) констант благоприятны для определения молекулярных весов растворенных веществ. Сама хлористая сера диссоциирована (вероятно, по схеме S2Сl2 + S2Cl2 Ы S2Сl+ + S2Сl3-) лишь ничтожно мало, но в ее растворах иногда происходит заметное образование солеобразных продуктов (по схемам, например, НgСl2 + S2Сl2Ы НgСl·S2Cl3- или S2СI2 + SbСl3Ы S2Сl·SbСl4-). Были получены и некоторые твердые сольваты (например, розовый 2СdО·S2Сl2 и серый Fе2(SO4)3·S2Сl2).
В резиновой промышленности монохлорид серы используется (как растворитель серы) при холодной вулканизации каучука, применяемой к различным мелким изделиям. Гораздо большее значение имеет горячая вулканизация, осуществляемая около 150 °С с помощью элементарной серы.
Сущность процесса вулканизации заключается главным образом в том, что атомы серы, соединяясь к нитевидным молекулам каучука по имеющимся в них двойным связям, как бы “сшивают” эти молекулы друг с другом. В результате вулканизации липкий и легко теряющий заданную форму сырой каучук превращается в упругую и эластичную резину.
Помимо рассмотренных выше галогенидных производных, для серы известны подобные по строению многосернистым водородам (т. е. содержащие цепи из атомов серы) галогенсульфаны общего типа SnГ2, где Г — Сl или Вr. Получают их обычно взаимодействием при низких температурах сульфанов с избытком галогенида серы или быстрым охлаждением продуктов взаимодействия при нагревании паров S2Г2 с водородом. Существовать могут, по-видимому, молекулы SnГ2 с очень большими значениями n. По мере роста этих значений теплоты образования хлорсульфанов последовательно снижаются (от 50 кДж/моль для S3Сl2, до 16,8 кДж/моль для S8Сl2). В индивидуальном состоянии были выделены члены ряда вплоть до S8Г2. Они представляют собой маслянистые жидкости различных оттенков оранжевого или красного цвета, обладающие неприятным запахом и в обычных условиях медленно разлагающиеся на S2Г2 и серу.
Конденсацией очень чистых хлорсульфанов SnСl2 и сульфанов Н2Sn в особых условиях (разбавленные растворы, отсутствие света) могут быть, по-видимому, синтезированы циклические молекулы элементарной серы различной атомности. Например, S6 образуется из Н2S и S2Сl2 по схеме:
НSН + СlSSСl + НSН + СlSSСl = 4 НCl + S6
Заметное взаимодействие серы с кислородом наступает лишь при повышенных температурах. Будучи подожжена на воздухе она сгорает синим пламенем с образованием диоксида по реакции:
S + O2 = SO2 + 297 кДж
Молекула О=S=O имеет структуру равнобедренного треугольника с атомом серы в вершине [d(SO) = 143 пм, Р(OSO) = 120°]. Диоксид серы (сернистый газ) представляет собой бесцветный газ с характерным резким запахом. Растворимость его весьма велика и составляет при обычных условиях около 40 объёмов на 1 объём воды.
Сернистый газ химически весьма активен. Характерные для него реакции можно разбить на три группы: а) протекающие без изменения валентности; б) связанные с её понижением; в) идущие с её повышением.
Процессом первого типа является прежде всего взаимодействие SO2 c водой, ведущее к образованию сернистой кислоты H2SO3. Последняя будучи кислотой средней силы, вместе с тем неустойчива, поэтому в её водном растворе имеют место равновесия:
H2O + SO2 Ы H2SO3Ы H+ + HSO3-Ы 2 H+ + SO32-
Постоянное наличие очень большой доли химически не связанного водой диоксида серы обуславливает резкий запах растворов сернистой кислоты. В свободном состоянии она не выделена.
При нагревании растворов сернистой кислоты диоксид серы улетучивается, вследствие чего приведённые выше равновесия смещаются влево. Напротив, при добавлении щелочей равновесия смещаются вправо (вследствие связывания ионов Н+), и жидкость, содержащая теперь уже соответствующие соли сернистой кислоты (сульфиты), перестает пахнуть диоксидом серы.
Будучи двухосновной, сернистая кислота дает два ряда солей: средние (сульфиты) и кислые (гидро- или бисульфиты). Подобно самим ионам SO32- и HSO3-, те и другие, как правило, бесцветны. Почти все бисульфиты устойчивы только в водных растворах, а из сульфитов хорошо растворимы главным образом сульфиты натрия и калия.
Из соединений серы с кислородом низший оксид — гемиоксид серы S2O — образуется при действии тлеющего электрического разряда на смесь SO2 с парами серы под уменьшенным давлением; реакция идёт по суммарной схеме
SO2 + 3 S = 2 S2O
Удобнее получать S2O (теплота образования из элементов 96 кДж/моль) проводимой при 160 °С и под давлением в 0,5 мм рт. ст. реакцией по схеме:
SOCl2 + Ag2S = 2 AgCl + S2O
То же соединение частично образуется при сжигании серы в токе кислорода под сильно уменьшенным давлением или при нагревании в вакууме тонко растёртой смеси CuO с серой (1:5 по массе). Ранее его считали монооксидом серы (SO или S2O2).
Молекула гемиоксида серы отвечает формуле S=S=O и имеет строение неравностороннего треугольника [d(SS) = 188, d(SO) = 146 пм, РSSO = 118°]. Она полярна (m = 1,47), причём средний атом, по-видимому, положителен по отношению к обоим крайним.
Гемиоксид серы представляет собой бурый газ, который может несколько часов сохраняться при комнатной температуре (в чистом и сухом сосуде) лишь под давлением не выше 40 мм рт. ст. При повышении концентрации быстро идёт реакция:
2 S2O = SO2 + 3 S
Сильное охлаждение переводит S2O в оранжево-красное твёрдое вещество, при нагревании до -30 °С распадающееся на SO2 и серу.
Молекулярным кислородом S2O при обычных условиях не окисляется, а водой легко разлагается, причём первичным продуктом гидролиза является H2S2O2, вступающая затем во вторичные реакции. Более или менее легко реагирует S2O с большинством металлов. При низких температурах для неё были получены некоторые продукты присоединения, например, жёлтый S2O·N(CH3)3.
Структурные параметры молекулы SO2 (m = 1,63) известны с очень большой точностью: d(SO) = 143,21 пм и РOSO = 119,536°. Приведённые числа наглядно показывают возможности использованного для их установления метода микроволновой спектроскопии. Энергия связи S=O оценивается в 535 кДж/моль.
Диоксид серы имеет точку плавления -75 °С (теплота плавления 7,5 кДж/моль) и точку кипения -10 °С (теплота испарения 25 кДж/моль). Критическая температура SO2 равна 157 °С при критическом давлении 78 атм. Термическая устойчивость SO2 весьма велика (по крайней мере до 2500 °С). Жидкий SO2 смешивается в любых соотношениях с рядом органических жидкостей (эфиром, бензолом, сероуглеродом и др.). Он является очень плохим проводником электрического тока. Наблюдающаяся ничтожная электропроводность обусловлена, вероятно, незначительной диссоциацией по схеме:
3 SO2 = S2O32+ + SO32-
Как растворитель диоксид серы обладает интересными особенностями. Например, галогеноводороды в нём практически нерастворимы, а свободный азот растворим довольно хорошо (причём с повышением температуры растворимость его возрастает). Элементарная сера в жидком SO2 нерастворима. Растворимость в ней воды довольно велика (около 1:5 по массе при обычных температурах), причём раствор содержит в основном индивидуальные молекулы H2O, а не их ассоциаты друг с другом или молекулами растворителя. По ряду Cl-Br-I растворимость галогенидов фосфора быстро уменьшается, а галогенидов натрия — быстро возрастает. Фториды лития и натрия (но не калия) растворимы лучше их хлоридов и даже бромидов. Хорошо растворим XeF4, причём образующийся бесцветный раствор не проводит электрический ток. Напротив, растворы солей в SO2 обычно имеют хорошую электропроводность (например, для NaBr при 0 °С имеем K = 5·10-5). Для некоторых из них были получены кристаллосольваты, например, жёлтый KI·(SO2)4. Подавляющее большинство солей растворимы в жидком SO2 крайне мало (менее 0,1 %). То же относится, по-видимому, и к свободным кислотам.
Уже при очень малых концентрациях диоксид серы создаёт неприятный вкус во рту и раздражает слизистые оболочки. Вдыхание воздуха, содержащего более 0,2 % SO2, вызывает хрипоту, одышку и быструю потерю сознания. Чувствительность отдельных людей к сернистому газу весьма различна, причём по мере привыкания к нему она заметно понижается. Хроническое отравление сернистым газом ведёт к потере аппетита, запорам и воспалению дыхательных путей. Максимально допустимой концентрацией SO2 в воздухе производственных помещений считается 0,01 мг/л. Содержащие SO2 баллоны должны иметь чёрную окраску с белой надписью “Сернистый ангидрид” и жёлтой чертой под ней.
Интересно отношение к диоксиду серы растительных организмов. Очень малое содержание его в воздухе (порядка 0,1 мг/м3), по-видимому, необходимо для нормального развития растений, тогда как более высокие концентрации оказываются очень вредными. Отдельные растения обладают разной чувствительностью по отношению к SO2. Так, из деревьев она наибольшая у ели и сосны, наименьшая — у берёзы и дуба. Из цветов особенно чувствительны к SO2 розы.
Помимо громадных количеств SO2, используемых для выработки серной кислоты, газ этот находит непосредственное применение в бумажном и текстильном производствах, при консервировании плодов и ягод, для предохранения вин от скисания (оптимальное содержание SO2 около 20 мг/л), дезинфекции помещений и т. д. Получают его для всех этих целей чаще всего сжиганием серы. Последняя загорается на воздухе около 300 °С.
Дезинфекция жилищ сернистым газом практиковалась ещё очень давно. Любопытно даваемое этому объяснение, которое приводит в своей книге Плиний: “Сера применяется для очищения жилищ, так как многие держатся мнения, что запах и горение серы могут предохранить от всяких чародейств и прогнать всякую нечистую силу”.
Давление диоксида серы над её водным раствором очень значительно, и содержание в нём недиссоциированных молекул H2SO3, по-видимому, невелико (для отношения [H2SO3]/[SO2] даётся значение 5·10-2). Путём охлаждения концентрированного раствора может быть выделен кристаллогидрат SO2·7H2O (т. пл. 12 °С), имеющий характер аддукта. При нагревании SO2 с водой до 150 °С в запаянной трубке происходит дисмутация по схеме:
3 SO2 + 2 H2O = 2 H2SO4 + S
Сернистая кислота характеризуется константами диссоциации К1 = 2·10-2 и К2 = 6·10-8. Для неё считают возможным существование двух структур:
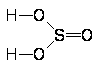 и
и 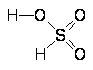 .
.
Какова структура самой кислоты, пока не ясно. Большинству её солей отвечает, по-видимому, первая из них, солям некоторых малоактивных металлов — вторая. Последняя вероятна также для кислых солей. Органические производные известны для обеих форм сернистой кислоты. Ион SO32- имеет структуру треугольной пирамиды с атомом S в вершине [d(SO) = 153 пм, РOSO = 105°].
Соли H2SO3 обычно получают взаимодействием SO2 с гидроксидами или карбонатами металлов в водной среде. Наибольшее практическое значение из них имеет известный только в растворе бисульфит кальция Ca(HSO3)2, который под названием “сульфитного щёлока” потребляется целлюлозной промышленностью для извлечения из древесины лигнина.
При кристаллизации раствора бисульфита натрия происходит отщепление воды по схеме:
2 NaHSO3 = H2O + Na2S2O5
с образованием натриевой соли неизвестной в свободном состоянии пиросернистой кислоты H2S2O5. Удобнее получать пиросульфит натрия сухим путём по реакции:
2 SO2 + 2 NaHCO3 = Na2S2O5 + 2 CO2 + H2O
Разложение его начинается уже при 100 °С и идёт в основном по схеме:
Na2S2O5 = Na2SO3 + SO2
Пиросульфит натрия находит применение в качестве консерванта влажного зерна и винограда. Аналогичная соль калия K2S2O5 может быть получена в виде твёрдых, бесцветных и лишь медленно растворяющихся в воде кристаллов путём насыщения сернистым газом раствора KHSO3. Под названием “метабисульфита” калия она применяется в фотографии и при крашении тканей. В её водных растворах имеет место равновесие
S2O52– + H2O Ы 2 HSO3–
c приблизительно равной концентрацией обоих ионов. Для калия, рубидия и цезия в твердом состоянии были получены не только пиросульфиты, но и бисульфиты.
При прокаливании сульфиты наиболее активных металлов разлагаются при температуре около 600 °С с образованием соответствующих солей серной и сероводородной кислот, например:
4 К2SO3 = 3 К2SO4 + К2S
Процесс этот аналогичен образованию перхлоратов и хлоридов при накаливании хлоратов.
Из процессов, не сопровождающихся изменением валентности серы практическое значение имеют реакции присоединения SO2, Н2SО3 и ее солей к некоторым органическим веществам. В частности, это относится ко многим красителям, причем образующиеся продукты присоединения большей частью либо бесцветны, либо слабоокрашены. На этом основано применение SО2 (обычно в растворе) для беления шерсти, шелка, соломы и т. п., т. е. таких материалов, которые не выдерживают отбелки при помощи сильных окислителей.
Значность серы не изменяется также при переходе от SO2 к галогенидным тионилам (SОГ2). Важнейшим из них является хлористый тионил, который может быть получен, например, по реакции:
SO3 + SCl2 Ы SOСl2 + SO2
Молекула OSCl2 полярна и имеет форму треугольной пирамиды с атомом серы в вершине [d(SO) = 144, d(SСl) = 208 пм, РOSСl = 107°, РСlSСl = 96°]. Хлористый тионил представляет собой бесцветную жидкость с резким запахом (т. пл. -100, т. кип. 76 °С). Нагревание выше температуры кипения ведет к его распаду на SO2, S2Cl2 и Сl2.
Жидкий SОСl2 имеет значение диэлектрической проницаемости e = 9 (при обычных температурах) и лишь в ничтожной степени диссоциирован (вероятно, по схеме: SОСl2 + SОСl2Ы SOС1++ SOСl3-). Он является плохим растворителем типичных солей, но хорошим для многих менее полярных веществ. При действии воды хлористый тионил нацело разлагается с образованием сернистой и соляной кислот: SOCl2 + 2 Н2О = Н2SО3 + 2 НСl. Он применяется при изготовлении красителей, фармацевтических препаратов и т. д. Им удобно пользоваться для получения безводных хлоридов металлов из их кристаллогидратов например, по схеме
СuСl2·2H2O + 2 SOCl2 = 4 НСl+ 2 SO2 + СuСl2
При длительном взаимодействии жидкого диоксида серы с фторидами Сs, Rb, K и Na(но не Li) по схеме МF + SО2 = МSО2F образуются соответствующие фторсульфинаты, по строению подобные хлоратам. Теплоты образования по приведенной реакции солей цезия, рубидия и калия равны соответственно 96, 88 и 75 кДж/моль. Свободная фторсульфиновая кислота (НSO2F) характеризуется точкой плавления -84 °C, но существует лишь в смеси жидких SO2 и НF (полностью смешивающихся друг с другом). При нагревании или под действием воды фторсульфинаты разлагаются.
Химические процессы, сопровождающиеся понижением валентности серы, для диоксида серы малохарактерны. Практически важно быстро идущее в присутствии катализатора (боксит) при 500 °С восстановление SO2 монооксидом углерода:
SO2 + 2 CO = 2 CO2 + S + 268 кДж
используемое иногда для извлечения серы из отходящих газов металлургических заводов.
Другим интересным методом является взаимодействие SO2 с сероводородом по уравнению:
SO2 + 2 H2S = 2 H2O + 3 S + 234 кДж
Реакция эта самопроизвольно протекает уже при обычных условиях, однако с заметной скоростью лишь в присутствии следов (т. е. очень малых количеств иода).
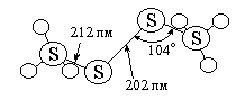
Рис. 8. Строение иона S4O62–.
В присутствии больших количеств воды взаимодействие SО2 и Н2S идет весьма сложно: кроме свободной серы образуется смесь сульфандисульфоновых кислот общей формулы Н2SnO6, обычно называемых политионовыми. Атомы серы в них непосредственно связаны друг с другом, образуя цепочку. В соответствии с этим, например, тетратионовой кислоте отвечает структурная формула НО-SО2-S-S-SO2-OH. Пространственное строение иона S4O62- показано на рис. 8.В водных растворах политионовые кислоты сильно диссоциированы и постепенно (при обычных температурах очень медленно) гидролитически разлагаются с образованием H2SO4, Н2SО3 и свободной серы. Сами по себе они совершенно неустойчивы, но их бесцветные диэфираты могут быть получены взаимодействием сульфанов с серным ангидридом в эфирной среде при -78 °С по общей схеме:
H2Sn + 2 SO3 +2 (С2Н5)2О = Н2Sn+2O6·2(С2H5)2O
В отличие от свободных кислот вполне устойчивы изученные лучше других солей политионаты калия (с n = 3ё6). Все они хорошо растворимы в воде (примерно 1: 4 по массе), но нерастворимы в спирте. При последовательном переходе n по ряду 3-6 устойчивость политионатов в кислой среде возрастает, а в щелочной среде быстро уменьшается.
Основные процессы взаимодействия SO2 и Н2S в водной среде могут быть описаны следующими схемами:
SO2 + 2 H2S = 2 H2O + 3 S SO2 + H2O + S = H2S2O3
3 SO2 + H2S = H2S4O6 3 SO2 + H2S + S = H2S5O6
В качестве первоначально возникающего промежуточного продукта принимают образование тиосернистой кислоты, которая может быть получена по схеме
Н2S + SO2 = H2S2O2
при -70 °С, но в обычных условиях разлагается на исходные газы. Обмен группами SО32- и S2O32-, по общей схеме
Н2SnO6 + Н2S2O3 Ы Н2Sn+1O6 + Н2SO3
может повести к образованию политионатов с различными значениями n. Следует отметить, что имеются и другие трактовки процесса образования политионовых кислот.
При получении растворов отдельных политионовых кислот исходят обычно из калийных солей, разлагая последние точно рассчитанным количеством какой-либо кислоты, образующей труднорастворимую соль калия (например, НСlO4). Общим исходным сырьем для получения самих политионатов калия служит его тиосульфат — К2S2О3. Реакции их образования протекают по уравнениям:
2 К2S2О3 + 3 SO2 = 2 К2S3О6 + S 2 K2S2O3 + I2 = К2S4O6 + 2 КI
2 К2S2О3 + SСl2 = К2S5О6 + 2KCl 2 К2S2О3 + S2Сl2=К2S6О6 + 2 КСl
Для получения тетра- и пентатионата калия удобнее исходить непосредственно из смеси политионовых кислот, образующейся в результате взаимодействия SO2 и Н2S. После отфильтровывания серы к жидкости добавляют СН3СООК и подвергают ее кристаллизации. При этом сначала выделяется К2S4O6 (призматические кристаллы), а затем К2S5О6 (кристаллы в форме табличек). Очень чистые соли при хранении устойчивы, тогда как загрязненные примесями разлагаются.
Высшие политионовые кислоты (с n > 6) менее устойчивы и хуже изучены. Для некоторых из них (например, Н2S18О6) были выделены соли сложных по составу объемистых катионов. Длина зигзагообразных цепей серы в политионовых кислотах, так же как в сульфанах и галогенсульфанах, принципиально не ограничена. Однако по мере возрастания n устойчивость их уменьшается.
Несколько особняком от рассмотренных выше политионовых кислот стоит дитионовая кислота (Н2S2O6), также известная лишь в растворе. Среди продуктов взаимодействия SO2 и Н2S она не содержится, получают же ее обычно исходя из марганцовой соли. Последняя образуется по уравнению
МnО2 + 2 SO2 = МnS2O6
при пропускании сернистого газа в воду, содержащую взвешенный гидрат диоксида марганца. После обменного разложения с Ва(ОН)2, и отделения осадка (BаSO3 и образующегося параллельно с основной реакцией ВаSO4) из раствора могут быть выделены бесцветные кристаллы ВаS2O6·2Н2О. Действуя на последнюю соль точно рассчитанным количеством Н2SO4, легко получить раствор свободной дитионовой кислоты. При попытке сильного концентрирования происходит ее распад по уравнению:
Н2S2O6 = H2SO4 + SO2
По отношению к окислителям дитионовая кислота значительно устойчивее остальных членов ряда. Соли ее большей частью хорошо кристаллизуются и все растворимы в воде. Строение дитионовой кислоты отвечает формуле НО-SО2-SO2-ОН с расстоянием d(SS) = 215 пм.
При обработке сернистым газом водной суспензии цинка по схеме:
Zn + 2 SO2 = ZnS2O4
образуется цинковая соль не выделенной в свободном состоянии дитионистой (иначе — гидросернистой) кислоты (Н2S2O4). После осаждения цинка при помощи Nа2СО3 и добавления к фильтрату NаСl выделяется бесцветный кристаллогидрат Nа2S2O4·2Н2О. Так как водная соль неустойчива, ее обезвоживают нагреванием со спиртом. Сравнительно устойчивый безводный дитионит (иначе — гидросульфит) натрия хорошо растворим в воде (1:5 по массе). Раствор сильно поглощает кислород и применяется при крашении тканей в качестве сильного восстановителя (окисляется до сульфита или сульфата). Дитионит натрия может быть получен также взаимодействием амальгамы натрия с насыщенным спиртовым раствором SO2 при температурах ниже 10 °С. Подобно Nа2S2O4, другие соли дитионистой кислоты и активных металлов бесцветны, хорошо кристаллизуются, легкорастворимы в воде (за исключением СаS2O4) и характеризуются сильными восстановительными свойствами. В растворах (и в виде кристаллогидратов) все они неустойчивы. Распад идет в основном по схеме:
2 S2O42- = S2O32- + S2О52-
Свободная H2S2O4 является кислотой средней силы (К1 = 0,5, К2 = 4·10-3) и крайне неустойчива — постепенно разлагается даже в разбавленных растворах и легко окисляется кислородом воздуха. По-видимому, как и в случае сернистой кислоты, возможно существование двух ее форм:
O O
кк зз
O=S-S=O O=S-S=O
Ѕ Ѕ Ѕ Ѕ
HO OH H H
Ион S2O42- слагается из двух групп SO2- соединенных, очень длинной — 239 пм — связью S-S.
Исходя из гидросульфита кобальта по реакции
СоS2O4 + 2 NаHCO3 = Nа2SO3 + СоSO2 + 2 СО2 + Н2О
может быть получен в виде бурого кристаллогидрата сульфоксилат кобальта СоSO2·3Н2О [вероятнее, Со2(SO2)2·6Н2О], являющийся солью сульфоксиловой кислоты (Н2SO2). Для последней возможны следующие структурные формулы:
HO-S-OH HO-S=O
Ѕ
H
Сама кислота образуется в качестве промежуточного продукта при гидролизе SСl2, и крайне неустойчива (распадается в основном по схеме 2Н2SО2 = Н2О + Н2S2O3). Существование других ее солей, помимо приведенной выше кобальтовой, с достоверностью не установлено.
Вместе с тем были получены некоторые органические производные сульфоксиловой кислоты. Из них кристаллическое соединение с формальдегидом состава NаНSO2·НСНО·2H2O (т. пл. 63 °С), известное под техническим названием «ронгалит», применяется в качестве сильного восстановителя (обычно — для снятия красителей с тканей). Вещество это хорошо растворимо в воде (1:2 по массе), устойчиво к действию щелочей и в нейтральной среде проявляет свои восстановительные свойства лишь около 100 °С. С точки зрения структуры ронгалит производится от второй из приведенных выше форм сульфоксиловой кислоты путем замещения гидроксильного водорода на натрий, а водорода, связанного с серой, на радикал СН2ОН.
Наиболее характерны для производных четырёхвалентной серы реакции (связанные с повышением её валентности: и сама сернистая кислота, и её соли являются сильными восстановителями. Растворы её уже при стоянии на воздухе постепенно (очень медленно) присоединяют кислород:
2 Na2SO3 + O2 = 2 Na2SO4
Несравненно быстрее (практически моментально) протекает окисление сернистой кислоты и сульфитов при действии таких окислителей, как КMnO4, Br2, I2 и т. п. В результате окисления образуется серная кислота или её соль.
Окисление растворов Н2SO3 и ее солей кислородом воздуха сопровождается ультрафиолетовым излучением с длиной волны около 220 нм. Данный процесс является, по-видимому, цепной реакцией, причем возникновение цепей связано с каталитическим влиянием примесей. В пользу этого говорит то обстоятельство, что скорость окисления тем меньше, чем тщательнее очищена взятая для растворения вода. Напротив, при прибавлении к раствору следов солей Fе, Сu и т. д. она сильно возрастает. С другой стороны, окисление сульфитов кислородом воздуха может быть сведено почти на нет добавкой к раствору незначительных количеств спирта, глицерина, SnСl2 и т. п. Задерживающее влияние небольших добавок некоторых веществ на окисление других кислородом воздуха носит название “антиокислительного катализа”. Явление это иногда практически используется в производстве органических препаратов.
Из отдельных процессов окисления сернистой кислоты заслуживает специального упоминания ее реакция с НIO3, позволяющая наглядно наблюдать зависимость скорости химического взаимодействия от концентрации и температуры. Суммарно она может быть выражена уравнением
НIO3 + 3 Н2SO3 = 3 Н2SO4 + НI
На самом деле реакция идет этим прямым путем лишь в первый момент, а после появления в растворе НI параллельно протекают следующие процессы:
HIO3 + 5 HI = 3 I2 + 3 H2O 3I2+3H2SO3+3H2O = 3 H2SO4 + 6 HI
Из них второй осуществляется быстрее первого. Поэтому иод может появиться в растворе лишь после полного окисления сернистой кислоты. Момент этот определяется по синему окрашиванию прибавляемого к смеси крахмала.
Рассматриваемая реакция интересна во многих отношениях. Прежде всего она дает пример протекающей в растворе цепной реакции: процесс по первому уравнению создает цепь, процессы по обоим следующим участвуют в ее развертывании. Затем она может служить примером аутокаталитической реакции, так как образующийся при ее протекании ион I’ ускоряет весь процесс (что легко установить, добавив к исходной смеси немного КI). Наконец, на ней же удобно проследить действие так называемых отрицательных катализаторов — веществ, добавка которых в небольших количествах существенно уменьшает скорость реакций. Таким веществом является в данном случае НgСl2, добавка которой к смеси заметно замедляет процесс.
Наряду с кислородом сульфиты способны присоединять также серу, переходя при этом в соли тиосерной кислоты, например по реакции:
Na2SO3 + S = Na2S2O3
Как и в случае кислорода, присоединение серы идёт медленно и для получения тиосульфатов приходится подвергать реакционную смесь кипячению.
Тиосерной кислоте отвечает формула:
 или
или 
Какая из них более правильна, пока неясно. И в той, и в другой молекуле атомы серы имеют разную степень окисления (+6 и -2). Это следует учитывать при составлении уравнений реакций, протекающих с участием Н2S2O3 и её солей.
По силе тиосерная кислота близка к серной, но в свободном состоянии она неустойчива и при выделении (путём подкисления растворов солей) распадается на сернистую кислоту и серу. Напротив, многие её соли (из которых известны лишь средние) устойчивы. Как правило они бесцветны и хорошо растворимы в воде. Наибольшее значение имеет тиосульфат натрия (гипосульфит) Na2S2O3·5H2O. Соль эта используется главным образом в фотографии и как сильный восстановитель, легко окисляющийся, например, по реакции:
Na2S2O3 + 4 Cl2 + 5 H2O = 2 H2SO4 + 2 NaCl + 6 HСl
Гипосульфит используется также в медицине.
Свободная серноватистая кислота может быть получена взаимодействием при -78 °С хлорсульфоновой кислоты с сероводородом по схеме:
НSО3С1 + Н2S = НСl + Н2S2O3
Она представляет собой маслянистую жидкость, при охлаждении жидким воздухом застывающую в стеклообразную массу. Заметное разложение серноватистой кислоты по схеме:
2 Н2S2O3 = Н2S + Н2S3О6
идет даже при -78 °С. Значительно устойчивее ее диэфират — Н2S2O3·2(С2Н5)2O, разлагающийся (на Н2S, SO3 и эфир) лишь выше -5 °С. Ион S2О32- имеет структуру тетраэдра с одним атомом серы около центра и другим на периферии [d(SS) = 148, d(SS) = 197 пм]. Результаты определения второй константы диссоциации серноватистой кислоты (К2 = 2·10-2) говорят о том, что она даже несколько сильнее серной (К2 = 1·10-2). При подкислении раствора Nа2S2O3 распад по схеме НS2O3’® НSО3’+ S наступает уже около рН = 4,5. Если раствор Na2S2O3 вводить по каплям в кипящую концентрированную НСl, происходит распад серноватистой кислоты по схеме:
H2S2О3 +Н2O = Н2S + Н2SО4
Интересен химизм получения гипосульфита путем взаимодействия серы со щелочью. Реакция эта в первой стадии протекает с одновременным окислением и восстановлением серы, т. е. аналогично соответствующим реакциям галогенов:
3 S + 6 NаОН = Nа2SO3 + 2 Nа2S + 3 Н2О
Находящаяся в избытке сера присоединяется затем к Nа2SO3, образуя гипосульфит. Параллельно происходит и образование полисульфидов натрия, сообщающих раствору желтый цвет. Для их разрушения с образованием гипосульфита в раствор пропускают SO2 до исчезновения желтой окраски жидкости.
Другой интересный способ получения гипосульфита основан на непосредственном взаимодействии SO2 и Н2S в щелочной среде. Если смесь обоих газов пропускать при сильном размешивании в раствор NаОН до его нейтрализации, то образуется гипосульфит:
4 SO2 + 2 Н2S + 6 NаОН = 3 Nа2S2O3 + 5 Н2О
При 48,5 °С гипосульфит плавится в своей кристаллизационной воде и около 100 °С теряет воду. Дальнейшее нагревание соли выше 220 °С сопровождается распадом, который идет в основном по схемам:
4 Nа2S2О3 = 3 Nа2SO4 + Nа2S5 Na2S5 = Nа2S + 4 S
Расплавленный гипосульфит очень склонен к переохлаждению. Иногда им пользуются как средой для определения молекулярных весов по понижению точки замерзания (криоскопическая константа 4,26 град). Из других тиосульфатов наиболее интересен малорастворимый (2 г/л) ВаS2O3, обменным разложением которого с сульфатами различных металлов удобно пользоваться для получения их тиосульфатов.
Подобно свободному хлору, другие сильные окислители (НОСl, Br2 и т. п.) окисляют гипосульфит до серной кислоты и ее солей. Иначе, а именно с образованием соли тетратионовой кислоты, протекает окисление гипосульфита сравнительно слабыми (или медленно действующими) окислителями, в частности иодом:
I2 +2 Nа2S2O3 = 2 NаI + Nа2S4O6
Эта реакция имеет большое значение для аналитической химии, так как служит основой одного из важнейших методов объемного анализа — так называемой иодометрии. Следует отметить, что в щелочной среде окисление гипосульфита иодом может протекать и до сульфата.
Медицинское применение гипосульфита довольно разнообразно. Его принимают внутрь (или вводят внутривенно) при отравлениях тяжелыми металлами, мышьяком и цианидами, а также для дезинфекции кишечника. Наружное (или внутреннее) применение показано при тяжелых ожогах и воспалениях кожи. Для лечения чесотки кожу больного повторно натирают концентрированным раствором Na2S2O3 и затем разбавленным раствором соляной кислоты.
Ближайшими аналогами Н2S2О3 являются политиосерные кислоты H2SnO3 (где n = 3ё7), которые могут быть получены в эфирной среде при -78 °С по общей схеме:
Н2Sn + SO3 + (С2Н5)2O = Н2Sn+1О3·(С2Н5)2O
Они хорошо растворимы в эфире и обладают сильно выраженными кислотными свойствами, но лишь одноосновны (в отличие от Н2S2О3), что соответствует строению сульфанмоносульфоновых кислот Н-Sn-1-SO2OН. И сами кислоты, и их соли разлагаются водой.
Для самого сернистого газа процессы, ведущие к повышению валентности серы, протекают значительно труднее, чем для сернистой кислоты и её солей. Наиболее важными из подобных реакций являются взаимодействия SO2 с хлором и кислородом.
С хлором диоксид серы непосредственно соединяется (на прямом солнечном свету) по реакции:
SO2 + Cl2 = SO2Cl2 + 92 кДж
Образующийся хлористый сульфурил представляет собой бесцветную жидкость с резким запахом. Холодная вода действует на него лишь медленно, но горячей он быстро разлагается с образованием серной и соляной кислот:
SO2Cl2 + 2 H2O = H2SO4 + 2 HСl
Вещество, дающее при взаимодействии с водой смесь галогенводородной и какой-либо другой кислоты, называется галогенангидридом последней. Хлористый сульфурил является, следовательно, хлорангидридом серной кислоты.
Если хлористый сульфурил можно рассматривать как серную кислоту, в которой на хлор заменены оба гидроксила, то продуктом подобного же замещения только одного из них является хлорсульфоновая кислота:
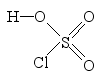
серная кислота хлорсульфоновая кислота хлористый сульфурил
Она может быть получена прямым соединением SO3 c HСl и, подобно хлористому сульфурилу, представляет собой бесцветную жидкость с резким запахом. Водой хлорсульфоновая кислота HSO3Cl бурно разлагается на HCl и H2SO4. Как и хлористый сульфурил, она находит применение при органических синтезах.
Хорошим катализатором при синтезе хлористого сульфурила (т. пл. -54, т. кип. 69 °С) является камфора. Молекула О2SСl2 полярна, ее пространственное строение отвечает неправильному тетраэдру с параметрами d(SO)) = 141 пм, РOSO = 123°, d(SСl) = 201 пм, РСlSСl = 100°. При нагревании выше 300 °С хлористый сульфурил распадается на SO2 и Сl2. Он является хорошим растворителем для SО3 и большинства хлоридов многовалентных металлов. Взаимодействие подобных растворов может служить удобным методом получения соответствующих безводных сульфатов. Известны также полисульфурилхлориды общей формулы SnO3n-1Cl2 с n = 2 ( т. пл. -37, т. кип. 152 °С), n = 3 (т. пл. -19 °С) и n = 4 (бесцветная жидкость).
Соответствующие оксохлоридам серы бромистые и иодистые производные неизвестны напротив, фтористые хорошо изучены. Фтористый сульфурил (SО2F2) образуется при непосредственном взаимодействии SО2 и F2, однако для начала реакции ей должен быть дан толчок (например, путем введения в смесь газов накаленной платиновой проволоки). Удобнее получать его действием фтора при температурах выше 100 °С на безводный сульфат натрия (реакция идет по уравнению
Nа2SO4 + 2 F2 = 2 NаF + SО2F2 + O2
Молекула О2SF2 полярна и имеет структуру тетраэдра с атомом серы около центра [d(SО) = 141 пм, РOSO = 124°, d(SF) = 153 пм, РFSF = 96°]. Фтористый сульфурил представляет собой бесцветный газ (т. пл. -136 °С, т. кип. -55 °С), химически довольно инертный. Так, он не взаимодействует ни с водой (даже при 140 °С), ни с расплавленным металлическим натрием. Однако растворами щелочей SО2F2 разлагается. Растворимость его в воде сравнительно невелика (1:10 по объему), в спирте — значительно больше. Термическое разложение SO2F2 начинается лишь выше 400 °С.
Известны также оксофториды: SOF4 (т. пл. -100, т. кип. -49 °С), (SF5)2O (т. пл. -115, т. кип. 31 °С), S2О5F2 (т. пл. -48, т. кип. 51 °С) и другие полисульфурилфториды, общего типа SО2F2·nSО3 (вплоть до n = 6), SO3F2 (т. пл. -158, т. кип. -31 °C), SOF6, (т. пл. -86, т. кип. -35 °С). Два последних являются производными фторноватистой кислоты, водород которой замещен на радикал SO2F или SF5. Для SF5OF даются следующие значения длин связей: d(SF) = 153, d(SO) = 164, d(ОF) = 143 пм. Интересен летучий сульфат (F5S)2SO4 (т. кип. 94 °С). Получены были и смешанные оксогалогениды: SO2FCl (т. пл. -125, т. кип. 7 °С), S2O5FCl (т. пл. -65, т. кип. 100 °C), SO2FBr (т. пл. -86, т. кип. 40 °С).
Молекула SOF4 по строению подобна SF4 (рис. 7), причем место свободной электронной пары занимает атом кислорода [d(SO) = 142, d(SF) = 155 (2) и 158 (2) пм, РFSF = 123° и 181°]. Контакт ОSF4 с избытком СsF при 100 °C в течение часа приводит к образованию СsОSF5. Характеристики этой интересной соли пока неизвестны. Под действием F2 она легко переходит в СsF и FОSF5. При 210 °С этот оксофторид разлагается на SF6 и кислород.
Хлорсульфоновая кислота (т. пл. -80 °С) малоустойчива и получить ее в чистом состоянии весьма трудно. Технический продукт обычно имеет темную окраску.
Соли хлорсульфоновой кислоты могут быть получены действием SО3 на растворы или взвеси хлоридов металлов в жидком SO2. Такая обработка взвесей NаСl и КСl дает бесцветные кристаллы NаС1·3SO3 и КСl·2SO3, которые после нагревания до 170 °С имеют состав МСl·SО3, т. е. представляют собой хлорсульфонаты МSО3С1. При добавлении жидкого SO3 к раствору АlСl3 в SO2 выпадают белые кристаллы АlСl3·3SO3, вероятно, представляющие собой хлорсульфонат алюминия — Аl(SO3Cl)3.
Аналогичная хлорсульфоновой, фторсульфоновая кислота представляет собой подвижную бесцветную жидкость (т. пл. -87, т. кип. 163'С), дымящую на воздухе. Она легко образуется при взаимодействии SО3 и НF (в газовой фазе НF + SО3 = HSO3F + 96 кДж), а водой медленно гидролизуется по схеме: SO2(ОН)F + Н2О Ы HF + H2SO4. Ввиду обратимости этой реакции (для ее константы равновесия дается значение 0,12) значительные количества HSO3F содержатся в смеси концентрированных HF и H2SO4. При полном отсутствии влаги фторсульфоновая кислота не действует на стекло и большинство металлов, но энергично реагирует со многими органическими веществами. Она растворяет AgF и фториды некоторых других элементов.
Кислотный характер выражен у фторсульфоновой кислоты сильнее, чем у серной. Для нее известны соли многих металлов, которые, как правило, легкорастворимы в воде (но растворимость СsSO3F при 0 °С равна лишь 0,1 моль/л). Получают их обычно прямым присоединением SO3 к соответствующим фторидам (при нагревании). Из них производные наиболее активных одновалентных металлов плавятся без разложения (например, КSО3F при 311 °С), тогда как фторсульфонат бария около 500 °С распадается по уравнению: Ва(SО3F)2 = ВаSO4 + SО2F2. Реакция эта может служить удобным методом получения фтористого сульфурила. Для иона SO3F- даются следующие значения длин связей: d(SO) = 143, d(SF) = 158 пм.
Из других фторсульфонатов следует прежде всего отметить FХеOSO2F, в кристалле которого d(FХе) = 194, d(ХеО) = 216, d(ОS) = 151, d(SO) = 142, d(SF) = = 153 пм, РFХеО = 178°, РХеОS = 123°. Интересны производные положительных галоидов, образующиеся при взаимодействии молекул Г2 с S2О6F2. Были получены черные ISО3F (т. пл. 52 °С) и I3SО3F (т. пл. 92 °С с разл.), жидкий при обычных условиях красновато-черный ВrSO3F (т. кип. 120 °С), светло-желтый СlSO3F (т. пл. -84, т. кип. 45 °С), оранжевый Вr(SO3F)3 (т. пл. 59 °С), желтые I(SO3F)3 (т. пл. 32 °С), IO2SO3F, IF3(SО3F)2, бесцветные КI(SO3F)4 и КВr(SO3F)4, К производным иода примыкают желтый нелетучий RеO3SO3F (т. пл. -33 °С). Интересны также имеющие ковалентный характер производные серы — SF4(ОSО2F)2, и SF4(OSF5)2. Сообщалось и о получении светло-желтого S4(SO3F)2. Все эти вещества гигроскопичны и тотчас разлагаются водой.
Фторсульфоновая кислота используется главным образом при органических синтезах. Взаимодействие ее с перхлоратом калия при нагревании по уравнению
КСIO4 + НSО3F = КНSО4 +FСlO3
может служить методом получения фторхлортриоксида.
Другой аналог НSO3Сl — бромсульфоновая кислота (НSO3Вг) была получена насыщением раствора SO3 в жидком SO2 сухим НВr при -35 °С. Она представляет собой бледно-желтое вещество, плавящееся около -7 °С с разложением (на Н2SO4, SO2 и Вr2).
Труднее, чем с хлором идёт соединение SO2 с кислородом, хотя сама по себе реакция экзотермична:
SO2 + O2 Ы 2 SO3 + 196 кДж.
Процесс с заметной скоростью пропекает только при достаточно высокой температурах и присутствии катализатора.
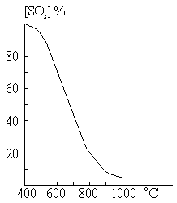
Рис. 9. Равновесие термической диссоциации SO3.
При быстром сгущении пара триоксида серы образуется бесцветная, похожая на лёд масса, которая затем медленно (быстрее под действием следов воды) переходит в белые шелковистые кристаллы. Обе модификации очень гигроскопичны и дымят на воздухе. Как видно из рис. 9, заметное разложение триоксида серы при нагревании (по реакции, обратимой её образованию) наступает лишь выше 400 °С.При сжигании серы на воздухе, наряду с SO2 образуется и SO3, но в него переходит менее 4 % от взятой серы. Помимо приведенной в основном тексте реакции, триоксид серы может быть получена термическим разложением Nа2S2O7 или безводного Fе2(SO4)3. Очень чистый SO3 образуется при взаимодействии SО2 с озоном.
Молекула SO3 неполярна и имеет структуру плоского треугольника с атомом серы в центре [d(SO) = 141 пм]. Для энергии и силовой константы связи S=O даются значения 472 кДж/моль.
Пар серного ангидрида (и раствор его в SO2) состоит преимущественно из молекул SO3, тогда как в жидкости преобладает смещенное вправо равновесие 3 SO3Ы (SO3)3. Молекулы тримера представляют собой кольца (рис. 10), образованные попеременно расположенными атомами S и O [со средними расстояниями d(S-O) = 161 и d(S=O) = 135 пм]. Из этих кольцевых молекул главным образом и состоит стекловидная a-форма серного ангидрида (т. пл. 17 °С). Основой структуры остальных его форм в твердом состоянии являются зигзагообразные цепи с более или менее значительным числом звеньев, -S(O2)-OS(O2)-O- (рис. 11), по-видимому, изолированные друг от друга у b-формы (т. пл. 32 °С) и спаявшимся в плоские сетки у g-формы (т. пл. 62 °С пол давл.) или в объемные структуры у d-формы (т. пл. 95 °С под давл.). Все модификации серного ангидрида обладают высоким давлением пара и легко возгоняются. Следует отметить, что получить различные формы твердого серного ангидрида в чистом виде весьма трудно и обычно приходится иметь дело с их смесями. Подобными смесями являются и продажные его препараты (белые кристаллы).
Рис. 10. Схема структуры (SO3)3. Рис. 11. Схема цепи (SO3)n.
Для хранения и транспортировки больших количеств наиболее удобна жидкая форма серного ангидрида (т. кип. 43 °С), которая сама по себе может быть устойчивой лишь при полном исключении даже ничтожных следов влаги. Стабилизация ее достигается введением специальных добавок (SOСl2, В2О3 и др.). При действии измельчённой серы на тщательно защищённый даже от следов воды жидкий SO3 осаждаются зеленовато-синие кристаллы сесквиоксида серы (S2O3). Оксид этот весьма неустойчив и сам по себе, а водой тотчас разлагается с выделением серы. Его самопроизвольный распад при обычных температурах идет в основном по схеме: 2 S2O3 = 3 SO2 + S. Строение его отвечает, вероятно, формуле OS=SO2.
Триоксид серы характеризуется сильными окислительными свойствами (восстанавливаясь обычно до SO2). С другой стороны он является кислотным ангидридом, причём образование H2SO4 из SO3 и воды сопровождается большим выделением тепла:
H2O + SO3 = H2SO4 + 63 кДж
Чистая 100 %-ная серная кислота (моногидрат) представляет собой бесцветную маслянистую жидкость, застывающую в кристаллическую массу при +10 °С. Реактивная серная кислота имеет обычно плотность 1,84 г, см3 и содержит около 95 % H2SO4. Затвердевает она лишь ниже -20 °С.
Рис. 12. Схема водородных связей в кристалле H2SO4.
Моногидрат может быть получен кристаллизацией концентрированной серной кислоты при -10 °С. Образующие его кристалл молекулы (НО)2SO2 соединены друг с другом довольно сильными (25 кДж/моль) водородными связями, как это схематически показано на рис. 12. Сама молекула (НО)2SO2 имеет структуру искаженного тетраэдра с атомом серы около центра и характеризуется следующими параметрами: (d(S-ОН) = 154 пм, РНО-S-ОН = 104°, d(S=O) = 143 пм, РOSO = 119°. В ионе HOSO3-, d(S-ОН) = 161 и d(SO) = 145 пм, а при переходе к иону SO42- тетраэдр приобретает правильную форму и параметры выравниваются [d(SO) = 148 пм].Температура плавления моногидрата равна 10,37 °С при теплоте плавления 10,5 кДж/моль. В обычных условиях он представляет собой очень вязкую жидкость с весьма высоким значением диэлектрической проницаемости (e = 100 при 25 °С). Незначительная собственная электролитическая диссоциация моногидрата протекает параллельно по двум направлениям: [Н3SO4+]·[НSO4-] = 2·10-4 и [Н3О+]·[НS2О7-] = 4·10-5. Его молекулярно-ионный состав может быть приближенно охарактеризован следующими данными (в %):
H2SO4 | HSO4- | H3SO4+ | H3O+ | HS2O7- | H2S2O7 |
| 99,5 | 0,18 | 0,14 | 0,09 | 0,05 | 0,04 |
При добавлении даже малых количеств воды преобладающей становится диссоциация по схеме:
Н2О + Н2SО4Ы Н3О+ + НSO4-
Моногидрат является ионизирующим растворителем, имеющим кислотный характер. В нём хорошо растворяются сульфаты многих металлов (переходя при этом в бисульфаты), тогда как соли других кислот растворяются, как правило, лишь при возможности их сольволиза (с переводом в бисульфаты). Азотная кислота ведет себя в моногидрате как слабое основание
HNO3 + 2 H2SO4Ы H3O+ + NO2+ + 2 HSO4–
хлорная — как очень слабая кислота
H2SO4 + HClO4 = H3SO4+ + ClO4–
Фторсульфоновая и хлорсульфоновая оказываются кислотами несколько более сильными (HSO3F > HSO3Cl > HClO4). Моногидрат хорошо растворяет многие органические вещества, имеющие в своём составе атомы с неподелёнными электронными парами (способными к присоединению протона). Некоторые из них могут быть затем выделены обратно в неизменённом состоянии путем простого разбавления раствора водой. Моногидрат обладает высоким значением криоскопической константы (6,12°) и им иногда пользуются как средой для определения молекулярных весов.
Концентрированная H2SO4 является довольно сильным окислителем, особенно при нагревании (восстанавливается обычно до SO2). Например, она окисляет HI и частично HВr (но не HСl) до свободных галогенов. Окисляются ею и многие металлы — Cu, Hg и др. (тогда как золото и платина по отношению к H2SO4 устойчивы). Так взаимодействие с медью идёт по уравнению:
Cu + 2 H2SO4 = CuSO4 + SO2+ H2O
Практически важно то обстоятельство, что очень крепкая (выше 75 %) серная кислота не действует на железо. Это позволяет хранить и перевозить её в стальных цистернах. Напротив, разбавленная H2SO4 легко растворяет железо с выделением водорода. Окислительные свойства для неё вовсе не характерны.
Действуя в качестве окислителя, серная кислота обычно восстанавливается до SO2. Однако наиболее сильными восстановителями она может быть восстановлена до S и даже H2S. С сероводородом концентрированная серная кислота реагирует по уравнению:
H2SO4 + H2S = 2H2O + SO2 + S
Следует отметить, что она частично восстанавливается также газообразным водородом и поэтому не может применяться для его осушки.
Крепкая серная кислота энергично поглощает влагу и поэтому часто применяется для осушки газов. От многих органических веществ, содержащих в своём составе водород и кислород, она отнимает воду, что нередко используется в технике. С этим же (а также с окислительными свойствами крепкой H2SO4) связано её разрушающее действие на растительные и животные ткани. Случайно попавшую при работе на кожу или платье серную кислоту следует тотчас же смыть большим количеством воды, затем смочить пострадавшее место разбавленным раствором аммиака и вновь промыть водой.
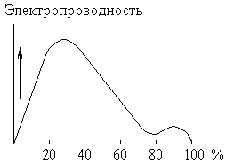
Рис. 13. Электропроводность растворов серной кислоты.
Растворение концентрированной серной кислоты в воде сопровождается значительным выделением тепла (и некоторым уменьшением общего объёма системы). Моногидрат почти не проводит электрического тока. Напротив, водные растворы серной кислоты являются хорошими проводниками. Как видно на рис. 13, максимальной электропроводностью обладает приблизительно 30 %-ная кислота. Минимум кривой соответствует гидрату состава H2SO4·H2O.Выделение тепла при растворении моногидрата в воде составляет (в зависимости от конечной концентрации раствора) до 84 кДж/моль H2SO4. Напротив, смешиванием 66 %-ной серной кислоты, предварительно охлажденной до 0 °С, со снегом (1:1 по массе) может быть достигнуто понижение температуры, до -37 °С.
Для серной кислоты известно несколько кристаллогидратов, состав которых показан на рис. 14. Из них наиболее бедный водой представляет собой соль оксония: H3O+HSO4–. Так как рассматриваемая система очень склонна к переохлаждению, фактически наблюдаемые в ней температуры замерзания лежат гораздо ниже температур плавления.
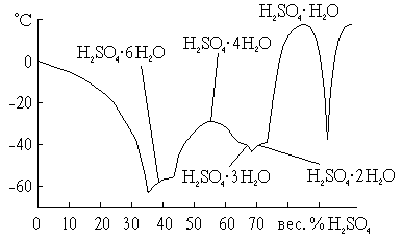
Рис. 14. Температуры плавления в системе H2O·H2SO4.
Изменение плотности водных растворов H2SO4 с её концентрацией (вес. %) дано ниже:
| 5 | 10 | 20 | 30 | 40 | 50 | 60 | |
15 °С | 1,033 | 1,068 | 1,142 | 1,222 | 1,307 | 1,399 | 1,502 |
25 °С | 1,030 | 1,064 | 1,137 | 1,215 | 1,299 | 1,391 | 1,494 |
| 70 | 80 | 90 | 95 | 97 | 100 | |
15 °С | 1,615 | 1,732 | 1,820 | 1,839 | 1,841 | 1,836 |
25 °С | 1,606 | 1,722 | 1,809 | 1,829 | 1,831 | 1,827 |
Как видно из этих данных, определение по плотности концентрации серной кислоты выше 90 вес. % становится весьма неточным.
Давление водяного пара над растворами H2SO4 различной концентрации при разных температурах показано на рис. 15. В качестве осушителя серная кислота может действовать лишь до тех пор, пока давление водяного пара над её раствором меньше, чем его парциальное давление в осушаемом газе.
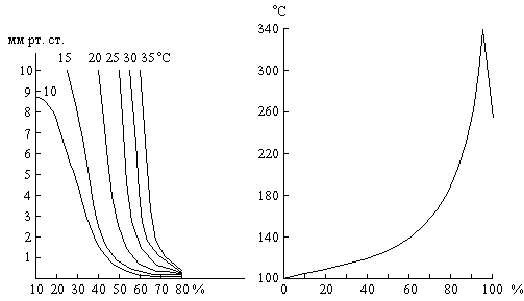
Рис. 15. Давление водяного пара Рис. 16. Температуры кипения над растворами H2SO4. растворов H2SO4.
При кипячении разбавленного раствора серной кислоты из него отгоняется вода, причём температура кипения повышается вплоть до 337 °С, когда начинает перегоняться 98,3 % H2SO4(рис. 16). Напротив, из более концентрированных растворов улетучивается избыток серного ангидрида. Пар кипящей при 337 °С серной кислоты частично диссоциирован на H2O и SO3, которые вновь соединяются при охлаждении. Высокая температура кипения серной кислоты позволяет использовать её для выделения при нагревании легколетучих кислот из их солей (например, HCl из NaCl).
Как сильная двухосновная кислота, H2SO4 даёт два ряда солей: средние (сульфаты) и кислые (гидросульфаты или бисульфаты), причём последние в твёрдом состоянии выделены лишь для немногих самых активных металлов (Na, K и др.). Большинство сернокислых солей бесцветно, хорошо кристаллизуется и легкорастворимо в воде. Из производных наиболее обычных металлов малорастворим CaSO4, ещё менее PbSO4 и практически нерастворим BaSO4.
По отношению к нагреванию сульфаты можно подразделить на две группы. Одни из них (например, соли Na, K, Ba) не разлагаются даже при 1000 °С, другие (например, соли Cu, Al, Fe) разлагаются на оксид металла и SO3 при гораздо более низких температурах. Некоторые, содержащие кристаллизационную воду сульфаты, иногда называют купоросами, например, CuSO4·5H2O — медный купорос, FeSO4·7H2O — железный купорос.
Как кислота H2SO4 в водных растворах близка по силе к хлорноватой в отношении первой стадии ионизации (H2SO4Ы H+ + HSO4–), но вторая стадия (HSO4–Ы H+ + SO42–) выражена гораздо слабее и характеризуется значением K2 = 1·10-2. Для зависимости этой константы от температуры имеются следующие данные:
Температура, °С | 5 | 15 | 25 | 35 | 100 |
K2 | 0,023 | 0,016 | 0,011 | 0,008 | 0,0008 |
Рис. 17. Характеристика сульфатного буфера.
В связи со сравнительно малым значением K2 растворы, содержащие смеси ионов HSO4– и SO42–, т. е. бисульфатов с сульфатами, обладают буферными свойствами. Общая характеристика такого сульфатного буфера дана на рис. 17. Нормальный раствор серной кислоты имеет pH = 0,3, децимолярный рН = 1,2 и сантинормальный рН = 2,1.Определения протонной активности (ан) в очень концентрированных растворах серной кислоты приводят к поразительным результатам. Так, для моногидрата она оказывается в 1010 раз большей, чем для молярного раствора. Это наводит на мысль о возможности существования в жидкостях при определенных условиях измеримых концентраций несольватированных протонов (Н+).
На разложении некоторых сернокислых солей при нагревании был основан способ получения серной кислоты, применявшийся алхимиками и затем вплоть до середины XVIII века. Исходным материалом служили природные минералы, содержащие сернокислые соли железа. Последние образовывались за счёт медленно протекавшего во влажном воздухе окисления сульфидов, например, по схеме:
4 FeS + 9 O2 + 2 H2O = 4 Fe(OH)SO4
При накаливании эта соль разлагалась с образованием монооксида железа, серной кислоты и серного ангидрида:
2 Fe(OH)SO4 = Fe2O3 + H2SO4 + SO3
Разбавляя продукт перегонки водой, получали серную кислоту желаемой концентрации.
Многие соли H2SO4 находят широкое техническое применение. Особенно велико оно для самой серной кислоты, громадные количества которой потребляются в промышленности — химической, нефтяной, металлургической и др.
Для промышленного получения серной кислоты применяются два метода: нитрозный и контактный. Основным исходным продуктом в обоих случаях является сернистый газ, получаемый сжиганием на воздухе серы (в США) или пирита — FeS2 ( в большинстве европейских стран, в том числе и России). Частично используется также SO2 отходящих газов, образующихся при выплавке металлов (Cu, Zn, Pb и др.) из их сернистых руд.
Для сжигания пирита (“серного колчедана”) на сернокислотных заводах пользуются специальными механизированными печами. Такая печь представляет собой цилиндр, разделённый по высоте на несколько соо6щающихся друг с другом отделений, в каждом из которых горящий FeS2 медленно перемешивается чугунными гребками, насажанными на общий вал. Своды отделений и гребки устроены так, что загружаемый сверху FeS2 последовательно проходит все отделения, после чего остатки от сгорания (“колчеданные огарки”) выбрасываются из печи. Воздух поступает в печь снизу и проходит вверх также через все отделения. Горение пирита идет по уравнению:
4 FeS2 + 11 O2 = 2 Fe2O3 + 8 SO2 + 3302 кДж
Температура в печи достигает 800 °С.
Обычный исходный газ сернокислотного производства содержит около 9 % SO2, 10 % O2 и 80 % N2. При использовании для обжига пирита воздуха, обогащенного кислородом, концентрация SO2 возрастает. Введение более богатых диоксидом серы газов в сернокислотное производство позволяет резко повысить выход серной кислоты.
Рис. 18. Схема печи для обжига пирита в кипящем слое.
Широко применяется для сжигания пирита (равно как и для проведения многих других технически важных процессов) метод “кипящего слоя”. Работа по этому методу осуществляется путём продувания подаваемой снизу сильной струи воздуха сквозь массу достаточно измельченного твердого вещества (или смеси веществ), Благодаря громадной общей реакционной поверхности поддерживаемых во взвешенном состоянии твёрдых частиц реакции протекают быстро и полностью. Печь для обжига пирита показана на рис. 18. Температура в кипящем слое поддерживается на уровне 900 °С; отходящий газ содержит до 14 % SO2.В качестве исходного сырья для производства серной кислоты может быть использован природный сульфат кальция. При сильном нагревании его в смеси с коксом протекает эндотермическая реакция:
CaSO4 + C + 392,92 кДж = CO + SO2 + CaO
Процесс проводят в цилиндрических вращающихся печах, прогреваемых сжиганием угольной пыли. Температура в печи достигает 1500 °С, отходящий газ содержит около 8 % SO2. Если в состав исходной шихты дополнительно вводить нужные количества глины и песка, то одновременно с каждой тонной вырабатываемой серной кислоты можно получать более тонны цемента.
Общий ежегодный вынос SO2 с отходящими газами металлургических заводов исчисляется многими миллионами тонн (из которых половина падает на серу). Газы эти содержат обычно не более 5 % SO2. Для извлечения из них диоксида серы предварительно охлаждённые и обеспыленные газы пропускают сквозь раствор смеси сульфита с бисульфитом, содержащий соли алюминия. Такой раствор хорошо поглощает диоксид серы на холоду и вновь выделяет её при нагревании. Роль солей алюминия сводится к повышению кислотности среды при нагревании за счёт резкого увеличения их гидролиза.
В основе другого метода улавливания диоксида серы из отходящих газов ( в том числе дымовых) лежит реакция
(NH4)2SO3 + SO2 + H2O Ы 2 NH4HSO3
равновесие которой смещается вправо на холоду и затем влево при нагревании под вакуумом. Следует отметить очень значительное поступление диоксида серы в атмосферу при сжигании содержащих серу топлив.
Рис. 19. Схема башенного метода получения серной кислоты.
Нитрозный метод получения H2SO4 был впервые применён в середине XVIII века. Его химическая сущность может быть выражена следующими реакциями:
I. SO2 + H2O + NO2 = H2SO4 + NO II. 2 NO + O2 = 2 NO2
Из первого уравнения видно, что являющийся окислителем диоксид азота NO2 восстанавливается до монооксида NO, а последний при взаимодействии с кислородом воздуха по второму уравнению вновь даёт диоксид. Таким образом, NO играет роль переносчика кислорода, т. е. по существу катализатором реакции окисления SO2 кислородом воздуха.
До 20-х годов текущего века процесс получения серной кислоты нитрозным методом проводился в больших свинцовых камерах (камерный метод). Теперь он осуществляется в специальных башнях (башенный способ). Получаемая по башенному способу кислота, как правило, содержит 76 % H2SO4 и несколько загрязнена различными примесями. Основным потребителем этой кислоты является промышленность минеральных удобрений.
Принципиальная схема башенного способа получения H2SO4 показана на рис. 19. Башни выкладываются из кислотоупорных керамических плит с наружным кожухом из листовой стали. Внутри они неплотно заполнены насадкой из кислотоупорной керамики. Поступающий из печи для сжигания пирита (А) газ освобождается от пыли в электрофильтре (Б) и затем подаётся в продукционные башни (В и Г), где встречается со стекающей сверху “нитрозой”, т. е. раствором оксидов азота в концентрированной серной кислоте. Раствор этот характеризуется следующими равновесиями:
NO + NO2 + 2 H2SO4 Ы N2O3 + H2SO4Ы 2 NOHSO4 + H2O
Таким образом, нитроза содержит оксиды азота и химически связанные (в виде HSO3NO2 — т. н. нитрозилсерной кислоты), и просто растворённые. Следует отметить, что окисление SO2 осуществляется только последними. При нагревании приведённые равновесия смещаются влево, при охлаждении — вправо.
В продукционных башнях, куда поступает горячий газ (а также подается вода), нитрозилсерная кислота полностью разлагается и происходит окисление практически всего вводимого сернистого газа. Готовая продукция отбирается из первой башни (В). В поглотительных башнях (Д и Е) происходит улавливание оксидов азота с образованием нитрозы, вновь подаваемой затем в продукционные башни. Выхлопные газы (свободный азот и др.) удаляются через верхнюю часть последней поглотительной башни (Е). Движение газов в системе поддерживается при помощи мощного вентилятора. Для компенсации некоторой потери оксидов азота в продукционные башни вводится азотная кислота.
Другой современный метод получения серной кислоты — контактный — освоен промышленностью лишь в конце прошлого столетия. Основой его является упоминавшаяся выше реакция:
2 SO2 + O2Ы 2 SO3 + 196 кДж
В присутствии платинового катализатора она около 400 °С протекает слева направо практически полностью. Образующийся SO3 улавливают крепкой серной кислотой. Стоимость производства по контактному способу несколько выше, чем по нитрозному, зато серная кислота получается сколь угодно крепкой и очень чистой. Последнее обусловлено тщательной предварительной очисткой образующихся при сжигании пирита газов, что необходимо для обеспечения нормальной работы катализатора. Основными потребителями контактной серной кислоты являются различные химические производства и нефтепромышленность (для очистки нефтепродуктов). Для контактного метода в общей продукции серной кислоты с каждым годом всё более возрастает.
Рис. 20. Схема контактного метода получения серной кислоты.
Рис. 21. Выход SO3 на катализаторах контактного метода.
Принципиальная схема получения серной кислоты контактным способом показана на рис. 20. Образующиеся в печи (А) газы последовательно проходят сквозь сухой электрофильтр (Б), увлажнительную башню (В), влажный электрофильтр (Г), осушительную башню (Д), содержащий катализатор окислительного процесса контактный аппарат (Е) и поглотительную башню (Ж). Из нижней части последней отбирается полученный олеум, а из верхней удаляются выхлопные газы (азот и др.). Большинство контактных заводов работает в настоящее время не с платиновыми, а со значительно более дешёвыми ванадиевыми катализаторами (V2O5, Ag3VO4 и др.), активность которых при реакции образования SO3 также весьма велика (рис. 21). Чаще всего применяют V2O5 с различными добавками (SiO2, KOH и др.).Растворы SO3 в серной кислоте дымят на воздухе вследствие выделения паров серного ангидрида. Поэтому содержащая растворённый SO3 серная кислота называется “дымящей” (иначе — “олеумом”). Так как H2SO4 растворяет серный ангидрид в любых соотношениях, выраженный формулой H2SO4·xSO3 состав олеума может быть различным. При х = 1, образуются бесцветные кристаллы пиросерной кислоты H2S2O7, строение которой сокращённо выражается формулой HO-SO2-O-SO2-OH. Соли её (пирофосфаты) могут быть получены нагреванием соответствующих бисульфатов, например, по реакции:
2 KHSO4 = H2O + K2S2O7
Они представляют собой бесцветные кристаллические вещества, под действием воды переходящие обратно в гидросульфаты.
В продажу олеум обычно поступает с содержанием растворённого SO3 не 6олее 25 %, т. е. значительно меньшим, чем то отвечает пиросерной кислоте (45 вес. %). Кристаллы последней плавятся при 35 °С и очень гигроскопичны. Накаливание пиросульфатов ведёт к отщеплению ими SO3, например, по схеме:
Na2S2O7 = Na2SO4 + SO3.
Были получены также соли (K, Na, NH4) фторопиросерной кислоты — HS2O6F.
При взаимодействии SO3 с HClO4 образуется, по-видимому, чрезвычайно взрывчатое соединение. Ему приписывается формула [ClO3+] [HS2O7–]. Если это действительно так, то пиросерная кислота оказывается даже сильнее хлорной.
Действием SO3 на Na2S2O7 может быть получен Na2S3O10. Соответствующей этой соли “трисерной” кислоте H2S3O10 отвечает структурная формула HO–SO2–O–SO2–O–SO2–OH. При обычных температурах Na2S3O10 устойчивее пиросульфата и переходит в него с отщеплением SO3 лишь выше 150 °С. У солей Ca, Sr и Ba такой переход происходит около 75 °С. Интересным производным H2S3O10 является красный взрывчатый (ClO2)2S3O10 (т. пл. 75 °С), который может быть получен взаимодействием KClO3 с избытком SO3. По-видимому, способны существовать также производные кислот H2S4O13 и H2S5O16. В самом олеуме предполагаются равновесия по общей схеме:
H2SO4 + n SO3 Ы H2Sn+1O3n+4 (где n = 1, 2 или 3).
При взаимодействии иода с олеумом образуются синие растворы. Результаты изучения их свойств говорят о наличии окислительно-восстановительного равновесия по схеме:
I2 + H2S2O7 + 3 SO3 Ы 2 I+ + 2 HS2O7– + SO2 или
2 I2 + H2S2O7 + 3 SO3 Ы 2 I2+ + 2 HS2O7– + SO2.
Вероятно, не исключена и возможность равновесия по схеме:
I2 + SO3 Ы I2SO3Ы I+ + ISO3–
Интересно, что цвет рассматриваемых растворов иода такой же, как и у его аддукта с крахмалом.
Если насыщенный раствор гидросульфата калия подвергнуть электролизу, то на катоде происходит выделение водорода и накопление КОН, а на аноде по схеме:
2 HSO4–- 2 e- = H2S2O8
образуется надсерная кислота. Получающийся в результате последующей нейтрализации H2S2O8 (персульфат калия) малорастворим и поэтому осаждается в виде бесцветных кристаллов. Большинство других солей надсерной кислоты хорошо растворимо в воде. Все персульфаты являются сильными окислителями. Например, серебро может быть окислено по уравнению:
(NH4)2S2O8 + 2 Ag = Ag2SO4 + (NH4)2SO4
Реакция эта используется в фотографии.

Рис. 22. Строение иона S2O82–.
Свободная надсерная кислота представляет собой бесцветные гигроскопичные кристаллы. Строение её молекулы выражается формулой HO-SO2-O-O-SO2-OH, т. е. она содержит пероксидную цепочку. Пространственная структура отвечающего ей иона S2O82- показана на рис. 22. Каждая половина этого рисунка в отдельности соответствует строению сульфат-иона.Хотя окислительные свойства надсерной кислоты (т. пл. 65 °С, с разложением) выражены очень сильно, однако со многими восстановителями она при обычных температурах реагирует в растворах настолько медленно, что окисление практически не происходит. Характерным для H2S2O8 катализатором, резко ускоряющим подобные процессы, является ион Ag2+. В его присутствии надсерная кислота способна окислять Mn2+ до HMnO4. Интересно, что тиосульфат окисляется ею только до тетратионата (а не до сульфата).
При взаимодействии H2S2O8 с концентрированным пероксидом водорода по уравнению
H2S2O8 + H2O2 = 2 H2SO5
образуется мононадсерная кислота, по строению отвечающая серной кислоте, в которой один гидроксид замещён на группу OOH. В свободном состоянии эта кислота представляет собой бесцветные и весьма гигроскопичные кристаллы (т. пл. 47 °С с разложением). Мононадсерная кислота является ещё более сильным окислителем, чем надсерная, и взаимодействие её со многими органическими веществами (например, бензолом) сопровождается взрывом. Она устойчивее H2S2O8 в кислых средах и менее устойчива в нейтральных и щелочных. Особенно это относится к pH = 9, когда раствор содержит равные концентрации ионов HSO5– и SO52–. Соли H2SO5 малоустойчивы. В них кислота обычно фигурирует как одноосновная. Обусловлено это затруднённостью ионизации водорода группы –OOH (K2 = 5·10-10).
Как следует из изложенного выше, обе надкислоты серы являются производными перекиси водорода. Сама H2O2 может быть легко получена из них, чем и пользуются в технике. Для этого подвергают электролизу концентрированную H2SO4, причём образующаяся вначале надсерная кислота быстро разлагается по реакции:
H2S2O8 + H2O = H2SO4 + H2SO5
Вслед за тем медленно разлагается и мононадсерная кислота:
H2SO5 + H2O = H2SO4 + H2O2
Образовавшуюся H2O2 отгоняют из реакционной смеси под уменьшенным давлением. Последняя из приведённых реакций заметно обратима, поэтому при смешивании концентрированных растворов H2O2 с концентрированной H2SO4 вновь образуется мононадсерная кислота.
В отличие от перекиси водорода обе надкислоты серы не реагируют с подкисленным раствором KMnO4 и при pH = 7,5–8,0 окисляют KI до свободного иода. Их взаимодействие с H2O2 сопровождается выделением кислорода. Мононадсерная кислота хорошо растворима в эфире, надсерная — плохо. Растворимость K2S2O8 в воде составляет при обычных условиях около 45 г/л, а (NH4)2S2O8 — более 750 г/л. Легкорастворим в воде также BaS2O8. При нагревании персульфаты с отщеплением кислорода переходят в пиросульфаты.
Длительным выдерживанием K2S2O8 над 65 %-ным олеумом может быть получен “надтетрасульфат” калия — K2S4O11. Эта бесцветная соль, сочетающая в себе особенности строения пиросульфата (связи S–O–S) и персульфата (связь S–O–O–S). Она энергично реагирует с водой, окисляет KI и обесцвечивает KMnO4). Потеря активного кислорода наступает при 130 °С, а около 250 °С начинается отщепление SO3.
Кроме надкислот были описаны пероксиды серы. При действии тлеющего электрического разряда на сильно охлаждаемую смесь SO3 с большим избытком кислорода образуется белое кристаллическое вещество, отвечающее формуле SO4 (молекулярный вес определён по понижению точки замерзания H2SO4). При 3 °С оно плавится и с частичным отщеплением кислорода переходит в маслянистую жидкость состава S2O7, затвердевающую при 0 °С. Водой SO4 разлагается с отщеплением кислорода лишь медленно, причём ни мононадсерная кислота, ни перекись водорода не образуются. Окислительные свойства SO4 (например, двухвалентный марганец переводится ею в семивалентный), судя по характеру протекания реакции, присущи не самому пероксиду серы, а выделяющемуся при его распаде атомарному кислороду. В растворе SO4 может быть, по-видимому, получен также действием фтора на концентрированную серную кислоту по реакции:
F2 + H2SO4 = 2 HF + SO4
Следует отметить, что в индивидуальной природе пероксидов серы и их точном соответствии приведенному выше составу нет уверенности.
Известны пероксофториды серы: S2O6F2 (т. пл. –55, т. кип. 67 °С) и S2O2F10 (т. пл. –95, т. кип. 49 °С), представляющие собой производные H2O2, в котором водороды замещены на радикалы –SO2F или –SF5. Первое из этих соединений довольно легко (с энергией активации 104,5 кДж/моль) диссоциирует по схеме:
S2O6F2 + 91,96 кДж Ы 2 SO3F
Ядерные расстояния в молекуле второго соединения составляют d(SF) = 156, d(OO) = 147 и d(SO) = 166 пм. Вещество это во многом похоже по свойствам на S2F10. Его термическая диссоциация начинается лишь около 200 °С. Описано также пероксидное производное состава S2O5F4 (т. пл. –95, т. кип. 35 °С), для которого вероятно наличие в молекуле пятичленного цикла. При облучении светом с длиной волны 365 нм смеси SO3 и F2O образуется желтовато-зеленый FSO2OOF (т. кип. 0 °С), устойчивый до 50 °С.
Круговорот серы в природе.
Из всех многообразных типов неорганических соединений серы, которые можно получить в лаборатории, лишь немногие способны к сколько-нибудь продолжительному существованию в природных условиях. Наряду с громадными количествами сульфатов и сульфидов только в сравнительно редких случаях встречаются залежи самородной серы и лишь как случайные и временные образования — сероводород и сернистый газ. Таким образом, неорганическая химия серы в земной коре и на её поверхности имеет в настоящее время дело почти исключительно с тремя типами соединений: H2SO4, H2S (включая их соли) и отчасти свободной S.
Ещё проще было, по-видимому, химическое состояние серы в эпоху формирования земной коры. Так как атмосфера тех времен свободного кислорода не содержала, выделяющийся из недр Земли сероводород не окислялся. Частично он образовывал соли с некоторыми металлами (Fe и др.) поверхностных пород земной коры, большей же частью находился в свободном состоянии.
Положение изменилось лишь после появления в атмосфере свободного кислорода. Как было сказано выше, сероводород легко окисляется с выделением серы. Процесс этот идёт и непосредственно на воздухе, но ещё быстрее под воздействием особого вида бактерий (серобактерий), получающих необходимую им для жизни энергию за счёт экзотермической реакции
2 H2S + O2 = 2 H2O + S + 531 кДж
Выделяющаяся сера откладывается в телах серобактерий, причём содержание её может доходить до 95 % их общей массы. Способствуя уничтожению вредного и для животных, и для растений сероводорода, эти бактерии играют важную положительную роль в живой природе.
Действие кислорода воздуха представляет собой основной природный процесс, ведущий к окислению сероводорода. Реакция иного типа протекает только в вулканических газах, где иногда выделяющийся H2S взаимодействует с одновременно выделяющимся SO2 по схеме:
2 H2S + SO2 = 2 H2O + 3 S
Дальнейшая судьба получающейся свободной серы зависит от наличия или отсутствия кислорода. Если сероводород выделяется на данном участке земной поверхности длительно и в значительных концентрациях, то постепенно накапливающаяся сера предохраняется его присутствием от дальнейшего окисления; в результате образуются более или менее мощные её залежи.
Напротив, избытком кислорода воздуха сера постепенно переводится в серную кислоту:
2 S + 3 O2 + 2 H2O = 2 H2SO4 + 1049 кДж
По этой же экзотермической реакции окисляется и сера, накопившаяся в организмах серобактерий, если последние попадают в среду, лишенную сероводорода.
При окислении серы первоначально должна была бы образовываться сернистая кислота. Между тем в природных условиях всегда получается серная. Это кажущееся противоречие объясняется тем, что из двух последовательных реакций
2 S + 2 O2 + 2 H2O = 2 H2SO3 + 656 кДж и
2 H2SO3 + O2 = 2 H2SO4 + 393 кДж
вторая протекает быстрее первой. Поэтому промежуточный продукт (H2SO3) и не накапливается.
Свободная H2SO4 встречается в природе крайне редко. Обычно тотчас же после своего образования она вступает в химическое взаимодействие с содержащимися в почве или воде солями более слабых кислот (главным образом углекислыми) и разлагает их по реакции, например:
CaCO3 + H2SO4 = CaSO4 + CO2 + H2O
Бульшая часть образующихся при этом сульфатов уносится водами рек, накапливается в морях и при их усыхании создаёт пласты различных сернокислых минералов (главным образом гипса — CaSO4·2H2O).
В противовес рассматривавшимся до сих пор окислительным процессам осуществляются и природные восстановительные. Из-за геологических смещений земной коры пласты сульфатов частично попадают в более глубокие слои Земли. Здесь под действием повышенной температуры они реагируют с увлечёнными при осаждении органическими веществами, например, по схеме (в качестве простейшего органического вещества взят метан):
CaSO4 + CH4 ® CaS + CO2 + 2 H2O ® CaCO3 + H2S + H2O.
Получающийся сероводород выходит на поверхность Земли либо прямо в газообразном состоянии, либо растворившись предварительно в подземных водах. Подобные сероводородные (“серные”) источники имеются в Пятигорске, Мацесте, Тбилиси и т. д. Водами этих источников широко пользуются в медицине при лечении различных заболеваний (кожных болезней, ревматизма и др.).
Аналогичные по химизму, но протекающие под влиянием сульфатовосстанавливающих бактерий процессы имеют место также в тех случаях, когда разложение органических веществ происходит под слоем воды, содержащей растворённые сульфаты. Такое сочетание условий характерно, в частности, для Черного моря, со дна которого вследствие этого всё время выделяется сероводород. Однако до верхних слоёв воды он не доходит, так как на глубине примерно 150 м встречается с проникающим сверху кислородом и окисляется им при содействии живущих на этом уровне серобактерий.
Другой восстановительный путь проходят сернокислые соли, задерживающиеся в почве. Извлекаемые из неё растениями сульфаты претерпевают затем сложные химические превращения, в результате которых образуются содержащие серу белковые вещества. Последние частично усваиваются животными. После отмирания животных и растительных организмов их белковые вещества разлагаются, причём сера выделяется в виде сероводорода, который таким образом вновь вводится в круговорот.
Таким образом, цикл превращений серы в природе представляет собой не просто круговорот, а вместе с тем определенный поступательный процесс, развивающийся в направлении перехода серы от более устойчивых при прежних условиях сульфидов к более устойчивым при современных условиях сульфатам.
45
12
ПОДГРУППА ТИТАНА.
На долю титана приходится около 0,2% от общего числа атомов земной коры, т.е. он является одним из весьма распростанённых в природе элементов. Доля циркония составляет 3·10-3 и гафния — 5·10-5%.
Хотя содержание в земной коре даже гафния больше, чем, например, иода или ртути, однако и титан и его аналоги ещё сравнительно плохо освоены практикой и иногда трактуются как “редкие” элементы. Обусловлено это их распылённостью, вследствие чего пригодные для промышленной разработки месторождения встречаются лишь в немногих местах земного шара. Другой важной причиной является трудность выделения рассматриваемых элементов из их природных соединений.
Цирконий открыт в 1789 г., титан — в 1791 г. Открытие гафния последовало лишь в 1923 г. Элемент № 104 был впервые (1964 г.) синтезирован Г. Н. Флёровым с сотрудниками. В СССР для него было предложено название курчатовий (Ku), в США — резерфордий (Rf). Известно несколько изотопов этого элемента, из которых наибольшей средней продолжительностью жизни атома (около 2 мин.) обладает 261Ku. На немногих атомах было показано, что с химической точки зрения курчатовий подобен гафнию.
Природный титан слагается из изотопов 46 (8,0), 47 (7,3), 48 (73,9), 49 (5,5), 50 (5,3%); цирконий — 90 (51,5%), 91 (11,2), 92 (17,1), 94 (17,4), 96 (2,8%); гафний — 174 (0,2), 176 (5,2), 177 (18,6), 178 (27,1), 179 (13,7), 180 (35,2%).
В основном состоянии атомы имеют строение внешних электронных оболочек 3d24s2 (Ti), 4d25s2 (Zr), 5d26s2 (Hf) и двухвалентны. Возбуждение четырёхвалентных состояний Тi (3d33s1), Zr (4d35s1), Hf (5d36s1) требует затраты соответственно 80, 59 и 167 кДж/моль, т.е. осуществляется гораздо легче, чем у элементов подгруппы германия.
Титан встречается в минералах ильменит (FeTiO3) и рутил (TiO2). Значительные количества титана содержат также некоторые железные руды, в частности уральские титаномагнетиты. Цирконий встречается главным образом в виде минералов цирконила (ZrSiO4) и бадделеита (ZrO2). Для гафния отдельные минералы пока не найдены. В виде примеси (порядка 1 атомн. %) он всегда содержится в рудах Zr.
Ничтожные количества титана постоянно содержатся в организмах растений и животных, но его биологическая роль не ясна. Титан и его аналоги не токсичны.
В свободном состоянии элементы подгруппы титана обычно получают путём восстановления их хлоридов магнием по схеме:
ЭCl4 + 2 Mg = 2 MgCl2 + Э.
Реакция проводится при нагревании исходных веществ до 900 °С в атмосфере аргона (под давлением).
Восстановление хлоридов титана и его аналогов магнием сопровождается значительным выделением тепла: 531 (Тi) и 322 (Zr) кДж/моль. Другим их восстановителем является металлический натрий, реакции с которым ещё более экзотермичны (приблизительно на 355 кДж/моль). Наиболее чистые образцы Ti, Zr и Hf были получены путём термического разложения на раскалённой вольфрамовой проволоке паров тетраиодидов под уменьшенным давлением.
По физическим свойствам элементы подгруппы титана являются типичными металлами, имеющими вид стали. Чистые металлы хорошо поддаются механической обработке. Но даже незначительные примеси некоторых элементов (Н, О, N, C и др.) сообщают им хрупкость. Их характерные константы:
| Ti | Zr | Hf | |
Плотность, г/см3 | 4,5 | 6,5 | 13,3 |
Температура плавления, °С | 1670 | 1855 | 2220 |
Температура кипения, °С | 3170 | 4330 | 5690 |
| Электропроводность (Нg = 1) | 2 | 2 | 3 |
В виде чистых компактных металлов все три элемента обладают высокой стойкостью по отношению к различным химическим воздействиям. Более реакционноспособны они в мелкораздробленном состоянии, при обычных температурах из всех кислот легко взаимодействуют лишь с HF. Лучшим растворителем для них является смесь плавиковой и азотной кислот, реагирующая по схеме:
3 Э + 18 НF + 4 HNO3 = 3 H2[ЭF6] + 4 NO + 8 H2O.
При высоких температурах Ti, Zr и Hf становятся химически очень активными. В этих условиях они энергично соединяются не только с галогенами, кислородом и серой, но также с углеродом и азотом. Порошки их способны поглощать большие количества водорода.
При общей высокой устойчивости чистых компактных металлов к различным химическим воздействиям элементы подгруппы титана проявляют и некоторые индивидуальные особенности. Так, по отношению к соляной или серной кислоте цирконий значительно устойчивее титана, а по отношению к влажному хлору или царской водке — наоборот. Под действием НF титан переходит в трёхвалентное состояние, а цирконий и гафний — в четырёхвалентное. При наличии ионов F- все три металла постепенно реагируют даже со слабыми кислотами. Концентрированной азотной кислотой титан (подобно олову) окисляется до нерастворимой титановой кислоты. В крепких растворах сильных щелочей порошок его растворяется с выделением водорода и образованием солей титановой кислоты. Цирконий и гафний по отношению к щелочам очень устойчивы.
Взаимодействие титана со фтором наступает уже около 150 °С, с другими газами — при 300-400 °С. В кислороде порошок титана загорается выше 500 °С, в азоте — выше 800 °С. Порошок циркония воспламеняется на воздухе уже около 250 °С. Сжиганием его в кислороде может быть получена температура до 4650 °С.
Обычно поверхность металлического циркония и титана покрыта очень тонкой, но плотной плёнкой оксида, полностью изолирующей металл от внешних воздействий. При некоторых условиях (например, при контакте Zr c очень влажным воздухом) плёнка может стать толстой, рыхлой и легко отделяющейся в результате того или иного случайного воздействия (например, сотрясения). Внезапно освобождённая от неё металлическая поверхность начинает энергично реагировать с кислородом и влагой воздуха, что иногда ведёт к самовозгоранию металла. Следует отметить, что горящий на воздухе цирконий потушить практически невозможно.
Каждый моль Ti, Zr или Hf способен сорбировать до 1 моля водорода, но быстро эта сорбция осуществляется лишь при высоких температурах (приблизительно с 400 для Тi и с 700 °С для Zr). Значительно легче устанавливается равновесие, если металл был предварительно прокалён в атмосфере Н2. Простейшим методом синтеза этих гидридов является достаточное нагревание и затем медленное охлаждение металла в атмосфере водорода под тем или иным его давлением. При малом содержании сорбированного водорода внешний вид металла существенно не изменяется, но при большем он превращается в серый или чёрный порошок (с плотностью 3,8 для ТiH2 и 5,5 г/см3 для ZrH2). Образование гидридов ЭН2 из элементов идёт с довольно значительным выделением тепла: около 125 (Тi) или 167 кДж/моль (Zr). В обычных условиях эти гидриды устойчивы на воздухе (но при поджигании загораются). Они довольно инертны также по отношению к большинству веществ, не являющихся сильными окислителями. Всё это указывает, как будто, на образование при сорбции водорода определённых химических соединений. Однако подобные соединения должны быть чрезвычайно неустойчивы, так как поглощённое металлом количество водорода меняется в зависимости от его давления и последовательно уменьшается при нагревании. Интересно, что образование гидрида титана наблюдалось также при длительном действии на металл крепкой соляной кислоты; основная реакция идёт по уравнению:
4 Тi + 6 HCl = 2 TiCl3 + 2 TiH2 + H2.
Гидрид титана является хорошим катализатором некоторых реакций гидрирования органических соединений. Он находит использование также в порошковой металлургии (как раскислитель). Гидрид циркония представляет интерес для ядерной энергетики (как замедлитесь нейтронов). Термическим разложением обоих гидридов могут быть получены тонкие плёнки соответствующего металла на различных материалах, что важно для ряда областей техники.
При нагревании Тi и Zr способны сорбировать также кислород (до 30 ат. %), причём поглощение сопровождается лишь очень небольшим увеличением объёма металлов. В меньших количествах они сорбируют и другие газы (N2 и пр.).
Практическое значение Ti и Zr особенно велико для металлургии специальных сталей. Оба металла используются и в качестве самостоятельных конструктивных материалов. Их соединения находят применение в различных отраслях промышленности. Гафний и его соединения пока почти не используются.
В металлургии титаном и цирконием пользуются в виде сплавов с железом — ферротитана и ферроциркония, содержащих 15-50% Тi или Zr. Выработка этих сплавов производится обычно путём прокаливания природных минералов Ti или Zr с углём в присутствии железной руды.
Добавка к стали уже 0,1% титана придаёт ей твёрдость и эластичность, что делает такую сталь очень хорошим материалом для изготовления рельсов, вагонных осей и колёс и т. д. Введением в сталь уже 0,1% Zr сильно повышает её твёрдость и вязкость, что особенно ценно для изготовления броневых плит и щитов. Как Тi, так и Zr нередко вводят также в различные сплавы Сu и Al.
Как конструкционный материал титан имеет очень благоприятное отношение прочности к массе в сочетании с высокой термической и коррозионной стойкостью. Он используется в самолетостроении. Цирконий (освобожденный от примеси гафния) является одним из важнейших конструкционных материалов при сооружении атомных реакторов. Порошок металлического циркония применяется иногда в составах для патронных запалов. Этот же порошок в смеси с нитратом циркония используется для изготовления световых сигналов, дающих при сгорании много света почти без дыма. Титан иногда применяется в качестве катализатора при различных реакциях, протекающих с участием свободного азота и водорода. При трении титана о стекло на последнем отлагается очень тонкая и плотная плёнка металла, что может быть использовано в электропромышленности для изготовления высокоомных сопротивлений. В обычных условиях титан не смачивается ртутью. Поверхность металлического циркония гидрофобна и не смачивается водой или водными растворами, что важно для конденсационных установок. Гафний находит использование главным образом в электронной технике (катоды телевизионных трубок и др.) и ядерной энергетике (как поглотитель нейтронов). Для пайки титана и циркония (в атмосфере аргона) наиболее пригодно чистое серебро.
Ежегодная мировая выработка титана оценивается сотнями тысяч тонн. Для циркония она имеет десяток тысяч, а для гафния — лишь сотню тонн.
В своих важнейших и наиболее характерных производных элементы подгруппы титана четырёхвалентны. Сам титан сравнительно легко образует малоустойчивые соединения, в которых он трёхвалентен. Производные двухвалентного титана немногочисленны и весьма неустойчивы. То же относится к производным трёх- и двухвалентного циркония и гафния, соединения которого по химическим свойствам очень близки к соответствующим соединениям циркония. Таким образом, по ряду Ti-Zr-Hf идёт понижение устойчивости низших валентностей, т.е. явление, обратное тому, которое имело место в подгруппе германия.
При нагревании элементов подгруппы титана в атмосфере кислорода они сгорают с образованием белых диоксидов (ЭО2). Последние очень тугоплавки и практически нерастворимы ни в воде, ни в разбавленных растворах кислот и щелочей. При нагревании с концентрированной серной кислотой они переходят в раствор лишь медленно, но легко могут быть переведены в растворимое состояние действием HF или сплавлением со щелочами. Диоксиды Ti и Zr находят разнообразное практическое применение. В частности, диоксид титана служит для изготовления очень хорошей белой краски (“титановые белила”).
Нагревание ТiO2 (т. пл. 1870 °С) выше 2200 °С ведёт к частичному отщеплению кислорода с образованием синего Тi3O5 (TiO2·Ti2O3) и затем тёмно-фиолетового Тi2O3. В стекольной промышленности диоксид титана применяется при изготовлении тугоплавких стёкол, в керамической — часто входит в состав эмалей, глазурей и фарфоровой массы. Искусственно получаемые в электрической печи прозрачные кристаллы рутила имеют показатель преломления (2,6), больший чем алмаз (2,4), и в шесть раз более высокую дисперсию света. Поэтому вырабатываемые из них драгоценные камни по красоте превосходят бриллианты. Диоксид титана служит хорошим катализатором при некоторых органических реакциях. Ежегодная мировая добыча TiO2 cоставляет около 1 млн. т.
Очень тугоплавкий (т. пл. 2850 °С) и в сплавленном состоянии чрезвычайно устойчивый по отношению к различным химическим воздействиям диоксид циркония применяется главным образом для изготовления огнеупорных изделий (тигли для плавки кварца и т. п.). Проводящие электрический ток путём переноса ионов О2- твёрдые растворы в ZrO2 некоторых других оксидов (например, Y2O3) используются как твёрдые электролиты при конструировании высокотемпературных топливных элементов. Введение ZrO2 в эмаль сообщает последней большую крепость и эластичность, а также некоторую устойчивость по отношению к температурным и химическим воздействиям. Содержащие ZrO2 cтёкла являются особенно устойчивыми по отношению к действию щелочей. Диоксид гафния более тугоплавкий (т. пл. 2900 °С), чем ZrO2.
Для всех элементов подгруппы титана были получены (взаимодействием элементов при нагревании) аналогичные по составу высшим оксидам сульфиды, селениды и теллуриды (кроме HfTe2). Цвет их коричневый (кроме жёлтого ТiS2 и чёрного ТiTe2). Термическая устойчивость этих веществ падает в ряду S-Se-Te, и при 600-700 °С могут проходить соответствующие реакции вытеснения. Опытами на спрессованных порошках установлено, что по рядам Ti-Zr-Hf и Te-Se-S уменьшается металлический и возрастает солеобразный характер соединений. Производные титана и ZrTe2 обладают металлической электропроводностью, ZrSe2, ZrS2, HfSe2 являются полупроводниками, а HfS2 — изолятором.
Довольно характерны для элементов подгруппы титана производные состава ЭХ3, где Х — S, Se, Te, которые могут быть получены непосредственным взаимодействием элементов при нагревании. По отношению к воздуху и воде они устойчивы, при нагревании на воздухе сгорают с образованием диоксидов входящих в них элементов. Хлор уже ниже 300 °С переводит их в соответствующие хлориды ЭСl4 (и SCl2, SeCl4 или TeCl4). Устойчивость этих веществ к действию концентрированной НСI уменьшается в ряду S-Se-Te. Крепким раствором NaOH или концентрированной Н2SO4 они при нагревании разлагаются, а в концентрированной HNO3 окисляются со взрывом.
При высоких температурах элементы подгруппы титана соединяются с углеродом, образуя карбиды типа ЭС. Реакции идут с выделением тепла: 192 (Ti), 200 (Zr) и 217 кДж/моль (Hf). Карбиды Ti, Zr и Hf представляют собой металлического вида кристаллы со структурой типа NaCl, очень твёрдые и тугоплавкие (т. пл. соответственно 3250, 3735 и 3890 °С). Сплав состава HfC·4TiC является самым тугоплавким из всех известных веществ (т. пл. 3990 °С). В противоположность карборунду, эти карбиды хорошо проводят электрический ток (лишь немногим хуже соответствующих металлов), с чем связано использование карбида титана при изготовлении дуговых ламп. Его часто вводят в состав керметов, используемых для изготовления разнообразных термостойких конструкций (лопаток газовых турбин и др.). Ввиду своей высокой твёрдости ТiC и ZrC иногда применяются в качестве шлифовального материала. При достаточном нагревании карбиды титана и его аналогов реагируют с галогенами, кислородом и азотом.
Близкородственны карбидам и похожи на них по свойствам силициды. Наиболее типичными формами для них являются ЭSi и ЭSi3.
При высоких температурах элементы соединяются с азотом. Получающиеся при этом металлического вида жёлтыенитриды Ti, Zr и Hf имеют состав, отвечающий формуле ЭN. Они образуются из элементов со значительным выделением тепла (соответственно 334, 364 и 368 кДж/моль) и представляют собой очень твёрдые, тугоплавкие (т. пл. 2930, 2950 и 2980 °С) и при обычных условиях химически инертные вещества, проводящие электрической ток значительно лучше соответствующих свободных металлов. Нагреванием до красного каления ZrCl4 в токе аммиака может быть получен коричневый нитрид состава Zr3N4 (промежуточными продуктами при этом являются ZrCl4·4NH3 и Zr(NH2)4). Титан в тех же условиях образует TiN, который ввиду своей чрезвычайной твёрдости применяется иногда (вместо алмазной пыли) для шлифовки драгоценных камней и т. д. Взаимодействие его с горячим раствором щёлочи протекает по уравнению:
2 TiN + 4 KOH + 2 H2O = 2 K2TiO3 + 2 NH3 + H2
Известен и двойной нитрид Li5TiN3, аналогичный производным кремния и германия.
Для фосфидов титана и его аналогов характерны типы Э2Р, ЭР и ЭР2. Это твёрдые серые вещества, термически устойчивые и не реагирующие с НСl, H2SO4 или HNO3 (но растворяющиеся в смеси HF + HNO3). Известны также двойные соединения состава Li5TiP3 и Li5TiAs3.
Отвечающие диоксидам ЭО2гидроксиды Э(ОН)4 могут быть получены действием щелочей на соединения типа ЭСl4. Они представляют собой студенистые осадки, почти нерастворимые в воде (но легко образующие коллоидные растворы). Гидрат диоксида титана имеет амфотерный характер, причём и основные, и особенно кислотные его свойства выражены весьма слабо. При переходе к Zr и Hf кислотные свойства ещё более ослабевают, а основные усиливаются. У гидроксидов Э(ОН)4 преобладают основные свойства, поэтому они растворяются в сильных кислотах, тогда как разбавленные щёлочи почти не действуют даже на Ti(OH)4.
Переход Zr(OH)4 (ПР = 1·10-54) и Hf(OH)4 к более бедной водой форме ЭО(ОН)2 осуществляется при 140 и 155 °С соответственно. Растворение обоих гидроксидов в крепких растворах сильных щелочей ведёт к образованию ионов Э(ОН)5’ или Э(ОН)6”; первый образуется при концентрации NaOH до 10 н., второй при более высокой. Из 15 н. раствора NaOH был выделен гафнат Na2[Hf(OH)6].
Соли гидратов диоксидов с металлами — титанаты, цирконаты и гафнаты получают сплавлением диоксидов с оксидами элементов или щелочами. Для образующихся солей наиболее характерны типы М2ЭО3 и М4ЭО4 (где М — одновалентный металл). Большинство их нерастворимо в воде, а растворимые подвергаются полному гидролизу.
Из титанатов, цирконатов и гафнатов наиболее интересен ВаТiO3. Соль эта является сегнетоэлектриком. Она обладает сверхвысокой диэлектрической проницаемостью в широком интервале температур (с максимумом при 120 °С). Сегнетоэлектрические свойства ВаTiO3 обусловлены возможностью смещения ионов Тi4+от средних положений в кристаллической решётке. Такое смещение ведёт к возникновению внутренних дипольных моментов, способных ориентироваться по внешнему полю.
Титанат бария используется для получения электрических конденсаторов исключительно большой ёмкости и генерации мощных ультразвуковых волн. В принципе, с его помощью механическая энергия (например, океанических волн) может быть непосредственно превращаема в электрическую.
Для элементов подгруппы титана характерны пероксидные соединения. Пероксид титана даже в ничтожных концентрациях сообщает водному раствору интенсивную жёлтую окраску. Его образованием (в сильнокислой среде) пользуются поэтому как чрезвычайно чувствительной реакцией и на титан и на пероксид водорода. Ответственным за окраску является ион TiO2••, содержащий пероксидную группу в трёхчленном цикле с титаном. Отвечающий ему сульфат был выделен в виде красного кристаллогидрата ТiO2SO4·3H2O. Связь между устойчивым в кислой среде пероксокатионом и устойчивым в щелочной среде пероксоанионом может быть представлена уравнением:
TiO22+ + 3 H2O2 = TiO84- + 6 H+.
Так как основные свойства гидроксидов ТiIV и его аналогов выражены сильнее кислотных, по отношению к воде соли бесцветных катионов Э4+ устойчивее титанатов, цирконатов и гафнатов. Но гидролиз этих солей очень значителен и даже в крепких растворах ведёт к образованию двухвалентных катионов титанила (TiO2+), цирконила (ZrO2+) и гафнила (HfO2+) по схеме:
Э4+ + Н2О = ЭО2+ + 2 Н+
Многие соли титана и его аналогов являются производными именно этих радикалов, а не Э4+. Таковы (TiO)SO4·2H2O, ЭОСl2·8H2O (где Э — Zr или Hf) и др. Дальнейший их гидролиз (особенно производных титана) идёт в меньшей, но всё же сильной степени.
Сульфат четырёхвалентного титана Ti(SO4)2 образуется при взаимодействии ТiCl4 c SO3 и SO2Cl2. Он представляет собой бесцветное, чрезвычайно гигроскопичное вещество. Его термическое разложение (в атмосфере сухого аргона) идёт с отщеплением SO3 и образованием ТiOSO4 (выше 150) или ТiO2 (выше 430 °С). В водной среде может быть получен только сульфат титанила — ТiOSO4·2H2O.
Сульфаты четырёхвалентных циркония и гафния известны и в безводном состоянии, и в виде кристаллогидратов Э(SO4)2·4H2O.
В образуемых сульфатами Тi, Zr и Hf комплексах с другими сульфатами координационное число центрального атома при переходе от Тi к Zr и Hf повышается. Так, комплексы типа М2Э(SO4)3 известны для всех трёх элементов, а типа М4Э(SO4)4 — только для циркония и гафния.
При одновременном наличии избытка КNCS сульфат титанила медленно растворяется в жидком аммиаке. Из образующегося красного раствора был выделен комплексный роданид состава К2[TiO (NCS)4]·2NH3, а действием на него КNH2 получен бурый амид титанила — ТiO(NH2)2, медленно гидролизующийся во влажном воздухе. Под действием избытка КNH2 он переходит в оранжево-коричневый ТiO(NHK)2 вспыхивающий при соприкосновении с воздухом и водой. Нагревание ТiO(NH2)2 сопровождается отщеплением аммиака и образованием сине-чёрного нитрида титанила — (TiO)3N2. Последний не взаимодействует с водой и разбавленными растворами кислот или щелочей, а при нагревании на воздухе переходит в TiO2.
Нитрат четырёхвалентного титана был получен при -80 °С по реакции:
TiCl4 + 4 ClNO3 = 4 Cl2 + Ti(NO3)4.
Он представляет собой бесцветное кристаллическое вещество (т. пл. 58 °С), в вакууме при 40 °С возгоняющееся. На воздухе нитрат титана разлагается с образованием белого оксонитрата ТiO(NO3)2, который при нагревании переходит в ТiO2. Оба соединения очень гигроскопичны и гидролитически разлагаются водой.
Безводные Zr(NO3)4 и ZrO(NO3)2 по большинству свойств аналогичны соответствующим производным титана. Однако в водных растворах нитраты циркония значительно устойчивее. Для них известны кристаллогидраты ZrO(NO3)2·2H2O и Zr(NO3)4·5H2O. Последняя соль легко отщепляет часть воды и переходит в нитрат цирконила. Для гафния известны кристаллогидраты HfO(NO3)2 с 2 и 6 молекулами воды и летучий аддукт HfO(NO3)2·N2O5.
Перхлораты ЭО(ClO4)2 известны в виде кристаллогидратов (с 6 Н2О для Ti и с 8 Н2О для Zr). Интересно, что соль титанила, плохо растворимая в воде, бензоле, ССI4 и диоксане, хорошо растворима в спирте и ацетоне.
Из производных других кислородных кислот для Zr и Hf особенно типичны гидрофосфаты Э(НРО4)2. Они отличаются тем, что практически нерастворимы в кислотах (кроме НF) и поэтому могут быть осаждены в сильнокислой среде. Это даёт возможность отделять Zr и Hf от всех других металлов (кроме Ра). Малорастворимы в кислотах и иодаты обоих металлов.
Для титана и его аналогов известны алкоголяты, образующиеся по схеме:
ЭСl4 + 4 ROH = 4 HCl + Э(OR)4.
Эти алкоголяты представляют собой жидкие или твёрдые летучие вещества, растворимые в бензоле, но гидролитически разлагающиеся водой. При растворении в соответствующих спиртах они способны образовывать комплексные кислоты типа Н2[Э(OR)6]. Интересно, что в твёрдом состоянии Тi(OC2H5)4 тетрамерен, а в бензольном растворе тримерен.
Из других производных Ti, Zr и Hf наибольшее значение имеют галогениды типа ЭГ4. Получают их обычно прокаливанием смеси диоксида элемента с углём в атмосфере галогена. Реакция идёт по схеме:
ЭО2 + 2 С + 2 Г2 = 2 СО + ЭГ4.
Характер галогенидов при переходе от Ti к Zr существенно изменяется. Так, TiCl4 представляет собой при обычных условиях жидкость, а ZrCl4 является типичной солью. За исключением ZrF4 (и HfF4) галонегиды ЭГ4 хорошо растворимы в воде.
Некоторые свойства галогенидов титана сопоставленны ниже:
TiF4 | TiCl4 | TiBr4 | TiI4 | |
| Теплота образования, кДж/моль | 1643 | 803 | 681 | 510 |
| Цвет | бесцв. | бесцв. | жёлт. | тёмно-красн. |
Температура плавления, °С | -23 | +39 | 159 | |
Температура кипения, °С | 283 | 136 | 231 | 377 |
Как растворитель неорганических соединений TiCl4 лучше всего растворяет вещества с типичной молекулярной структурой. Растворимость в нём солеобразных соединений, как правило, тем выше, чем больше размеры аниона.
При постепенном добавлении TiCl4 к жидкому аммиаку образуется жёлтый осадок аммиаката ТiCl4·6NH3. В действительности он представляет собой смесь состава Ti(NH2)3Cl + 3NH4Cl, так как при отмывании его жидким аммиаком NH4Cl удаляется и остаётся красный Ti(NH2)3Cl. Нагревание последнего в вакууме сопровождается отщеплением NH3 с образованием в остатке зеленовато-голубого нитрохлорида NTiCl. Продуктами термического разложения аммиакатов TiBr4 и TiI4 являются соответственно NTiBr и NTiI. Последний выше 400 °С переходит в ТiN. Взаимодействие Ti(NO3)4 с KNH2 в жидком аммиаке ведёт к образованию коричневого Ti(NH2)4, который легко переходит во взрывчатый Ti(NH)2.
Гидролиз галогенидов ЭГ4 протекает в основном по схемам:
ZrГ4 + Н2О Ы ZrOГ2 + 2 HГ и
TiГ4 + 2 H2O Ы TiO2 + 4 HГ.
Образующийся в результате гидролиза гидрат диоксида титана начинает осаждаться уже при рН = 1,5. Исключением являются фториды, образующие с водой комплексные кислоты типа Н2[ЭОF4] и поэтому почти не подвергающиеся гидролизу даже при нагревании растворов, из которых могут выделяться кристаллогидраты TiF4·2H2O и ZrF4·3H2O. В последнем из них установлено наличие димерных молекул с фторидными мостиками по типу F3ZrFFZrF3.
Оксохлориды циркония и гафния выделены из раствора в виде кристаллогидратов ЭОСI2·2Н2О. Около 150 °С хлорид цирконила обезвоживается, а выше 250 °С разлагается по схеме:
2 ZrOCl2 = ZrCl4 + ZrO2.
Прямым синтезом безводный хлорид цирконила был получен при -15°С в ССI4 по схеме:
ZrCl4 + Cl2O = 2 Cl2 + ZrOCl2.
Он представляет собой белое кристаллическое вещество, нерастворимое в неполярных растворителях и сильно гидролизуемое водой. Его кристаллическая решётка слагается из полимеризованных путём образования связей -Zr-O-Zr-O- анионов [ZrOCl4]2-и катионов [ZrO]2+.
Весьма характерным свойством большинства галогенидов ЭГ4 является их сильно выраженная склонность к реакциям присоединения. Общим примером для всех трёх элементов подгруппы титана могут служить жёлтые (Ti) или бесцветные (Zr, Hf) двойные соединения состава ЭCl4·POCl3 и ЭCl4·2POCl3 плавящиеся соответственно при 104 и 105 °С (Ti), 205 и 185 °С (Zr) или 222 и 198 °С (Hf).
Для всех рассматриваемых соединений очень характерно комплексообразование с соответствующими галогеноводородными кислотами и особенно с их солями. Наиболее типичны комплексные производные с общей формулой М2ЭГ6 (где М — одновалентный металл). Они хорошо кристаллизуются и подвергаются гидролизу гораздо менее, чем исходные галогениды ЭГ4. Это указывает на устойчивость комплексных ионов ЭГ6” в растворе.
В то время как почти все комплексные соли Zr и Hf бесцветны, окраска производных титана сильно зависит от природы входящего в них галогена:
| Комплексная кислота | H2[TiF6] | H2[TiCl6] | H2[TiBr6] | H2[TiI6] |
| Цвет солей | бесцветный | жёлтый | красный | тёмно-красный |
Устойчивость солей комплексных кислот типа Н2ЭГ6, в общем, возрастает по ряду Ti-Zr-Hf и уменьшается в ряду галогенов F-Cl-Br-I.
Производные трёхвалентных элементов более или менее характерны лишь для титана. Тёмно-фиолетовый оксид Тi2O3 (т. пл. 1820 °С) может быть получен прокаливанием TiO2 до 1200 °C в токе водорода. В качестве промежуточного продукта при 700-1000 °С образуется синий Ti2O3.
В воде Ti2O3 практически нерастворим. Его гидроксид образуется в виде тёмно-коричневого осадка при действии щелочей на растворы солей трёхвалентного титана. Он начинает осаждаться из кислых растворов при рН = 4, имеет только основные свойства и в избытке щелочи не растворяется. Однако производящиеся от HTiO2 титаниты металлов (Li, Na, Mg, Mn) были получены сухим путём. Известна также сине-чёрная “титановая бронза” состава Na0,2TiO2.
Гидроксид титана (III) легко окисляется кислородом воздуха. Если в растворе нет других способных окисляться веществ, одновременно с окислением Ti(OH)3 идёт образование пероксида водорода. В присутствии Са(ОН)2 (связывающего Н2О2) реакция протекает по уравнению:
2 Ti(ОН)3 + O2 + 2 H2O = 2 Ti(OH)4 + H2O2.
Азотнокислые соли Тi(OH)3 восстанавливает до аммиака.
Фиолетовый порошок ТiCl3 может быть получен пропусканием смеси паров ТiCl4 c избытком водорода сквозь нагретую до 650 °С трубку. Нагревание вызывает его возгонку (с частичным образованием димерных молекул Ti2Cl6) и затем дисмутацию по схеме:
2 TiCl3 = TiCl4 + TiCl2.
Интересно, что уже при обычных условиях тетрахлорид титана постепенно восстанавливается металлической медью, образуя чёрное соединение состава CuTiCl4 (т. е. СuCl·TiCl3).
Трёххлористый титан образуется также при действии на TiCl4 водорода в момент выделения (Zn + кислота). При этом бесцветный раствор окрашивается в характерный для ионов Ti3+ фиолетовый цвет, и из него может быть выделен кристаллогидрат состава ТiCl3·6H2O. Известен и малоустойчивый зелёный кристаллогидрат того же состава, выделяющийся из насыщенного HCl раствора TiCl3. Структуре обеих форм, равно как и аналогичных кристаллогидратов СrCl3, отвечают формулы [Э(ОН2)6]Cl3 и [Э(ОН2)4Cl2]Cl·2Н2О. При стоянии в открытом сосуде раствор TiCl3 постепенно обесцвечивается ввиду окисления Ti3+ до Ti4+ кислородом воздуха по реакции:
4 TiCl3 + O2 +2 H2O = 4 TiOCl2 + 4 HCl.
Ион Тi3+ является одним из очень немногих восстановителей, довольно быстро восстанавливающих (в кислой среде) перхлораты до хлоридов. В присутствии платины Тi3+ окисляется водой (с выделением водорода).
Безводный Ti2(SO4)3 имеет зелёный цвет. В воде он нерастворим, а раствор его в разбавленной серной кислоте имеет обычную для солей Ti3+ фиолетовую окраску. От сульфата трёхвалентного титана производятся комплексные соли, главным образом типов М[Ti(SO4)2]·12H2O (где М — Сs или Rb) и M[Ti3(SO4)5] (с переменным в зависимости от природы катиона содержанием кристаллизационной воды).
Соединения двухвалентных элементов подгруппы титана плохо изучены. Теплота образования TiO (т. пл. 1750 °С) составляет 518 кДж/моль. Он получается в виде золотисто-жёлтой компактной массы нагреванием в вакууме до 1700 °С спрессованной смеси TiO2 + Ti. Интересным способом его образования является термическое разложение (в высоком вакууме при 1000 °С) нитрила титанила. Похожий по виду на металл, тёмно-коричневый TiS получен прокаливанием TiS2 в токе водорода (первоначально при этом образуются сульфиды промежуточного состава, в частности Ti2S3). Известны также TiSe, TiTe и силицид состава Ti2Si.
Все ЭГ2 (равно как и чёрный самовоспламеняющийся на воздухе НfBr2) образуются при нагревании соответствующих галогенидов ЭГ3 без доступа воздуха за счёт их разложения по схеме:
2 ЭГ3 = ЭГ4 + ЭГ2.
При несколько более высоких температурах галогениды ЭГ2 сами подвергаются дисмутации по схеме: 2 ЭГ2 = ЭГ4 + Э (например, дисмутация ZrI3 идёт при 310, аZrI2 — при 430 °С).
Двухлористый титан может быть получен также восстановлением TiCl4 водородом при 700 °С. Он хорошо растворим в воде (и спирте), а с жидким аммиаком даёт серый аммиакат TiCl2·4NH3. Раствор TiCl2 может быть получен восстановлением TiCl4 амальгамой натрия. В результате окисления кислородом воздуха бесцветный раствор TiCl2 быстро буреет, затем становится фиолетовым (Ti3+) и, наконец, вновь обесцвечивается (Ti4+). Получаемый действием щёлочи на раствор TiCl2 чёрный осадок Ti(OH)2 исключительно легко окисляется.
Подгруппа селена.
По электронным структурам нейтральных атомов селен и теллур являются прямыми аналогами серы. Эти три элемента, вместе взятые, иногда называют халькогенами (“рождающими медь”). Наиболее тяжелый элемент подгруппы — полоний — радиоактивен, относится к наименее распространенным (содержание в земной коре около 2·10-15 %) и сравнительно с другими мало изучен.
Теллур открыт в 1798 г., селен — в 1817 г., полоний — в 1898 г.
Природный селен состоит из изотопов с массовыми числами 74 (0,9 %), 76 (9,0 %), 77 (7,6 %), 78 (23,5 %), 80 (49,8 %), 82 (9,2 %), а теллур — из изотопов с массовыми числами 120 (0,1 %), 122 (2,4 %), 123 (0,9%), 124 (4,6 %), 125 (7,0 %), 126 (18,7 %), 128 (31,8 %), 130 (34,5 %). Приведённые данные показывают, что у обоих элементов количественно преобладают более тяжёлые разновидности атомов. Для полония известны только радиоактивные изотопы, из которых в природе встречается 210Po (средняя продолжительность жизни атома 200 дней).
По структуре внешних электронных слоев атомы селена (4s24p4), теллура (5s25р4) и полония (6s26p4) подобны атому серы и в своем основном состоянии тоже двухвалентны. Возбуждение четырехвалентного состояния требует довольно большой затраты энергии.
Источником полония ранее служили радионосные руды. В настоящее время его получают искусственно (исходя из висмута). Элементарный полоний может быть выделен из растворов его соединений с помощью электролиза (в ряду напряжений он располагается между медью и серебром). При изучении этого элемента исследованию обычно подвергаются лишь миллиграммовые количества, что обусловлено даже не столько трудностью его получения, сколько очень сильной радиоактивностью полония (в темноте можно видеть его светло-голубое самосвечение).
Содержание селена в земной коре составляет 1·10-5 %, теллура — 1·10-7 %. Для обоих элементов наиболее характерно совместное нахождение с такими металлами, как Cu, Pb, Hg, Ag и Au. Самостоятельно минералы Se и Te встречаются крайне редко, обычно же лишь в виде примесей к аналогичным минералам серы.
Основным источником промышленного получения селена и теллура служат осадки (“шламы”), образующиеся при электролитической выработке меди. Ежегодная мировая добыча селена имеет порядок 1 тыс. т, теллура — 200 т.
Извлечение Sе и Те из производственных отходов металлургической (или сернокислотной) промышленности основано на переводе обоих элементов в четырёхвалентное состояние с последующим их восстановлением сернистым газом. Восстановление первоначально ведется в крепкой (10-12 н) соляной кислоте, причём выделяется только селен. Затем, после сильного разбавления жидкости водой, выделяется теллур.
Очистка селена от примесей может быть проведена различными методами. Например, можно воспользоваться его хорошей растворимостью в горячем концентрированном растворе Nа2SО3. Если затем добавить немного раствора Аl2(SO4)3, то выпадающий осадок гидроксида (и основных солей) алюминия увлекает с собой примеси к исходному селену. Отфильтровав этот осадок, раствор затем охлаждают, что сопровождается выделением очищенного селена. Очистить последний можно также путем продувания при 450 °С струи воздуха сквозь его расплав с последующей перегонкой остатка в вакууме.
Теллур очищают перегонкой в вакууме или в токе водорода. Для его очистки пользуются также переводом теллура в основную азотнокислую соль состава Те2О3(OH)NО3 с последующим обратным выделением после очистки этой соли перекристаллизацией. Комбинированием всех трёх приемов может быть достигнута очень хорошая очистка.
Селен применяется главным образом в полупроводниковой технике (изготовление выпрямителей переменного тока и т. д.). Он используется также в стекольной промышленности, при вулканизации каучука, в фотографии и при изготовлении некоторых оптических и сигнальных приборов. Последнее применение основано на том, что электропроводность селена сильно возрастает с увеличением интенсивности его освещения. По своей спектральной характеристике селеновый фотоэлемент довольно близок к человеческому глазу, но гораздо чувствительнее.
Этим свойством в некоторой степени обладает и теллур, электропроводность которого резко возрастает также при высоких давлениях (в 100 раз при 12 тыс. атм и становится металической при 30 тыс. атм). Потребляется он главным образом в производстве свинцовых кабелей: добавка теллура (до 0,1 %) к свинцу сильно повышает его твёрдость и эластичность. Такой свинец оказывается также более стойким по отношению к химическим воздействиям. Кроме того, теллур находит применение при изготовлении полупроводников и при вулканизации каучука. Соединения его используются для окраски стекла и фарфора, в фотографии и микробиологии (для окрашивания микробов).
При выделении из растворов своих соединений оба элемента осаждаются в виде порошков, соответственно красного и коричневого цвета. Однако наиболее типичны для них те модификации, некоторые свойства которых сопоставлены ниже с соответствующими свойствами кислорода серы и полония.
| Элемент | При обычных условиях | Температура плавления, | Температура кипения, | Плотность в твёрдом | |
| Агрегатное состояние | Цвет | °С | °С | состоянии, г/см3 | |
| O | газ | бесцв. | –218 | –183 | 1,3 |
| S | твёрд. | жёлтый | 119 | 445 | 2,1 |
| Se | » | серый | 221 | 685 | 4,8 |
| Te | » | серебристо-белый | 450 | 990 | 6,2 |
| Po | » | » » | 254 | 962 | 9,3 |
Селен и теллур устойчивы на воздухе и нерастворимы в воде. Все соединения селена сильно ядовиты.
Основные аллотропические модификации селена можно свести к трем формам, обладающим различной внутренней структурой. Самой устойчивой из них является серый селен, образованный бесконечными спиральными цепями его атомов [d(SеSе) = 232 пм, РSеSeSе = 105°] уложенными в кристалле параллельно друг другу. Две другие формы по отношению к этой метастабильны. Из них красный селен в двух своих кристаллических разновидностях (Sеa и Sеb) образован кольцевыми молекулами Sе8 со средними параметрами d(SеSе) = 235 пм и Рa = 106°. Третья форма — амфотерный селен (порошкообразный или стекловидный) — образована зигзагообразными цепями, перепутанными друг с другом. При обычных температурах метастабильные формы селена в стабильную (серую) практически не переходят. Серый селен является полупроводником р-типа с шириной запрещенной зоны 1,5 эВ.
Выделяемый действием сернистого газа при получении селена его кирпично-красный порошок настолько тонок, что лишь с трудом оседает. Около 50 °С он темнеет и спекается в почти чёрную хрупкую массу стекловидного селена (плотность 4,3 г/см3). Последний может быть получен также быстрым охлаждением расплавленного селена (например, выливанием его в воду). После такой “закалки” масса долгое время сохраняет пластичное состояние. Уже при 50 °С твердый стеклообразный селен начинает размягчаться, а около 100 °С претерпевает протекающее со значительным выделением тепла (около 46 кДж/моль) и кратковременным разжижением массы превращение в серую форму.
При контакте стекловидного селена с некоторыми органическими жидкостями (СS2 и др.) он медленно в темноте и быстрее на свету переходит в красный кристаллический селен. Последний несколько растворим в сероуглероде (около 0,05 % при обычных условиях и 0,1 % при 46 °С). Упариванием такого раствора ниже 72 °С могут быть получены моноклинные кристаллы Sе (пл. 4,5 г/см3), а выше этой температуры — гексагональные кристаллы Sе (пл. 4,4 г/см3). При быстром нагревании до 180 °С красный селен плавится без изменения, вообще же переход его в серую форму начинает протекать уже выше 110 °С.
Стабильная серая форма может быть получена также из расплавленного селена, но лишь при условии его очень медленного охлаждения. Удобнее её получать возгонкой селена под уменьшенным давлением. При нагревании выше 72 °С селен становится пластичным и легко поддается механическим деформациям. С повышением давления его температура плавления возрастает, достигая при 4 тыс. атм примерно 270 °С. Плавление сопровождается резким увеличением объёма (приблизительно на 16 %). Теплота плавления селена составляет 6,7 кДж/моль. В отличие от серы вязкость коричнево-красной жидкости (плотность около 4,05 г/см3) с повышением температуры непрерывно уменьшается. Теплота испарения селена равна 29,3 кДж/моль. В его желтоватых парах имеет место равновесие 1/4 Sе8Ы 1/3 Sе6Ы 1/2 Sе4Ы Sе2, смещённое вправо более, чем у серы.
Обе основные формы теллура — порошкообразная тёмно-коричневая (“аморфная”) и металлоподобная серебристо-белая — слагаются из бесконечных спиральных цепей его атомов [d(ТеТе) = 286 пм, РТеТеТе = 102°]. Переход коричневой формы в металлоподобную (похожую по внешнему виду на олово, но хрупкую и имеющую полупроводниковые свойства) осуществляется с заметной скоростью только при нагревании (теплота перехода около 0,8 кДж/моль). Работа выхода электрона из металлоподобной формы равна 4,7 эВ. Под высокими давлениями существуют аллотропические модификации теллура, природа которых пока не изучена.
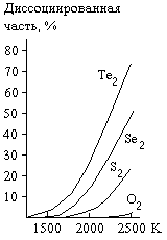
Рис. 1. Диаграмма состояния Рис. 2. Термическая диссоциация
теллура при высоких давлениях молекул Э2.
(тыс. атм).
При повышенных температурах теллур настолько пластичен, что поддаётся прессованию. В вакууме он легко возгоняется. Теплота его плавления равна 17,6, а испарения — 51 кДж/моль. Плавление сопровождается увеличением объема приблизительно на 5 %. Интересной особенностью жидкого теллура является наличие у него максимума плотности немного выше температуры плавления (как у воды). С жидким иодом он смешивается в любых соотношениях. Золотисто-жёлтые пары теллура состоят преимущественно из молекул Те2.
Полоний имеет уже явно выраженный вид металла. По физическим свойствам он более похож на Т1, Вi и Рb, чем на Те (напротив, по химическим — чрезвычайно похож на теллур). Для полония известны две аллотропические формы, переходящие друг в друга при разных температурах (a ® b при 54 °С и b ® a при 18 °С) и сосуществующие в этом температурном интервале. Следует отметить, что данные эти не очень надежны из-за саморазогревания полония, обусловленного его сильной радиоактивностью.
Как видно из рис. 2, термическая диссоциация молекул Sе2 и Те2 осуществляется значительно легче, чем в случаях серы и кислорода. Последнее связано с общим характером изменения ядерных расстояний и энергий диссоциации по ряду О - Те:
| Молекулы | О2 | S2 | Se2 | Te2 |
| d, пм | 121 | 189 | 219 | 257 |
| Энергия диссоциации, кДж/моль | 497 | 418 | 318 | 217 |
Теплоты атомизации селена и теллура при 25 °С равны соответственно 226 и 192 кДж/моль.
Друг с другом селен и теллур не соединяются (но смешанные кристаллы образуют). Селен способен соединяться с серой, давая циклические смешанные молекулы SnSе8-n и образованные ими кристаллы, примерами которых могут служить красные Sе4S4, (т. пл. 113 °C) и светло-оранжевые Sе2S6 (т. пл. 122 °С). Для теллура было получено соединение с серой состава ТеS7.
Теллур уже при обычных условиях очень медленно взаимодействует с водой по схеме:
Те + 2 Н2О = ТеО2 + 2 Н2
При нагревании подобным же образом реагирует с водой и аморфный селен, тогда как кристаллический не взаимодействует с ней даже при 150 °С. Оба элемента растворяются в растворах Nа2Sn с образованием ионов SеS22- или ТеS32-. Mелкораздробленные Sе и Те растворяются в холодной концентрированной серной кислоте. Установлено, что растворение идет с образованием желтых ионов Sе42+, зеленых Sе82+ или красных Те42+. При разбавлении раствора водой происходит обратное выделение Sе или Те. Из раствора теллура в жидком серном ангидриде избыток SO3, может быть удалён нагреванием. Дальнейшее нагревание полученного таким путем ТеSO4, сопровождается отщеплением SO2 и образованием черного твердого вещества, по составу отвечающего формуле ТеО (но представляющего собой, по-видимому, смесь ТеО2+ Те). Полоний ведет себя аналогично теллуру, давая красный РоSO3 и черный РоО.
Повышенное содержание селена в почвах может обусловить частичное замещение им серы при построении белковых молекул растительных организмов. Результатом являются заболевания как самих растений, так и питающихся ими животных: у скота выпадает шерсть, размягчаются копыта и т. д. Отмечалось избирательное накопление селена мухоморами.
При приеме внутрь соединения селена действуют подобно мышьяку. После отравлений ими появляется очень неприятный запах от всего тела и выдыхаемого воздуха. Газообразные производные селена уже в ничтожных концентрациях вызывают головную боль, раздражение верхних дыхательных путей, продолжительную потерю обоняния и затяжной насморк. При попадании его соединений на кожу образуются сыпи и болезненные воспаления. Вместе с тем ничтожные дозы селенитов (порядка 3 мкг на 100 г пищи), по-видимому, предотвращают заболевания некротического характера. Отмечалось также прямая связь между остротой зрения животных и содержанием селена в сетчатке их глаз.
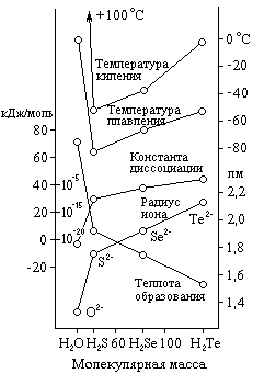
Рис. 3. Свойства водородных соединений элементов VI группы.
Соединения теллура значительно менее ядовиты. В организме они быстро восстанавливаются до элементарного Те, который затем медленно выделяется в виде постепенно образующихся органических производных, обладающих сильнейшим чесночным запасом. Последний появляется после поступления в организм даже ничтожных количеств Те (десятимиллионных долей грамма). Известен случай, когда работавшим с теллуристыми соединениями химикам пришлось на несколько недель выселиться из города, так как исходивший от них запах был невыносим для окружающих. При лечении селеновых и теллуровых отравлений рекомендуется принимать в повышенных дозах витамин С (аскорбиновую кислоту).Недавно проведенными исследованиями установлено постоянное наличие следов полония в табачном дыме. Оказалось, что с дымом от пачки сигарет человек поглощает такую дозу облучения a-частицами, которая в 4 раза превышает признаваемую безопасной по международному соглашению. Анализ мочи курильщиков показал, что она содержит полония в 6 раз больше, чем у некурящих. Это значит, что полоний циркулирует по организму и может накапливаться (как и продукт его распада — свинец) не только в легких, но и в любом другом органе. Предполагается, что статистически установленная значительно большая частота заболеваний раком курильщиков по сравнению с некурящими обусловлена главным образом именно радиоактивностью полония.
С химической стороны селен и теллур в общем похожи на серу. Из неметаллов они наиболее энергично взаимодействуют с фтором и хлором, а с кислородом соединяются лишь после предварительного нагревания. С газообразным водородом частично реагирует при повышенных температурах только селен, тогда как теллур с ним непосредственно не соединяется. Со многими металлами Se и Te дают при нагревании аналогичные сульфидам селениды и теллуриды (например, K2Se, K2Te).
Действием на них разбавленных кислот могут быть получены селеноводород (H2Se) и теллуроводород (H2Te). Оба они при обычных условиях представляют собой бесцветные газы с неприятными запахами. Растворимость их в воде примерно такая же, как у сероводорода, причем растворы показывают ясно выраженную кислую реакцию. Некоторые свойства рассматриваемых соединений сопоставлены в приводимой ниже таблице и на рис. 3 с аналогичными свойствами H2O и H2S. Для приблизительной ориентировки в размерах соответствующих молекул приведены также радиусы ионов Э2–.
| Соединения | Теплота образования, кДж/моль | Температура плавления, °С | Температура кипения, °С | Константа диссоциации K1 | Радиус иона Э2–, пм |
H2O | 68,5 | 100 | 2·10-16 | 1,32 | |
H2S | 5 | –86 | –60 | 1·10-7 | 1,74 |
H2Se | –8 | –66 | –41 | 1·10-4 | 1,91 |
H2Te | –24 | –51 | –2 | 2·10-3 | 2,11 |
Из данных таблицы видно, что H2Se и H2Te являются кислотами более сильными, чем, например, уксусная (К = 2·10-5). По отношению к нагреванию чистый H2Se довольно устойчив, тогда как H2Te легко разлагается на элементы. Кислородом воздуха оба соединения постепенно окисляются и в газообразном состоянии и особенно в растворе уже при обычных температурах. В общем, восстановительные свойства характерны для H2Se и H2Te еще более, чем для сероводорода. Гидрид полония (H2Po) крайне неустойчив.
Отвечающие типичным валентным переходам селена и теллура окислительно-восстановительные потенциалы сопоставлены ниже (верхняя цифра относится к кислой среде, нижняя — к щелочной):
-2 0 +4 +6
Sе -0,40 +0,74 +1,15
-0,92 -0,37 +0,05
Те -0,72 +0.53 +1,02
-1,14 -0,57 +0,4
Для полония наиболее типична валентность +4, менее характерны -2 (полониды) и +2 (известны, в частности, черный РоS и красный РоSO3). Существование валентности +6 установлено, но отвечающие ей производные пока не выделены.
Помимо разложения селенидов, Н2Sе может быть получен пропусканием тока водорода над нагретым до 600 °С селеном. Удобнее получать его нагреванием селена с парафином или действием воды на Аl2Sе3. Образование Н2Те хорошо идет при электролизе сильно охлажденных растворов кислот с теллуровым катодом. Получить его можно также действием 4 н. НСl на Аl2Те3. Термический распад Н2Sе с заметной скоростью идет лишь выше 300 °С, тогда как Н2Те постепенно разлагается уже при обычных температурах. Селенистый водород гораздо более ядовит, чем сероводород. Растворимость его в воде при обычных условиях составляет около 3:1 по объему. Взаимодействие Н2Sе с серой медленно протекает по схеме:
S + Н2Sе = Н2S + Sе
(т. е. аналогично реакции Сl2 + 2 НВr = 2 НСl + Вr2).
По строению молекул Н2Sе и Н2Те подобны Н2S. Для селенистого водорода d(SеН) = 146 пм, РНSеН = 91°. Молекула теллуристого водорода изучена хуже. Для нее d(ТеН) = 169 пм, РНТеН = 89,5°. Средние энергии связей Sе-Н и Те-Н оцениваются соответственно в 313,5 и 263,3 кДж/моль.
Для селеноводорода (К2 = 1·10-11) известны два ряда солей — кислые и средние, для теллуроводорода (К2 = 1·10-5) — лишь средние. Из них производные наиболее активных одновалентных металлов бесцветны и легкорастворимы в воде. Некоторые относящиеся сюда соли были выделены и в форме кристаллогидратов (например, Nа2Те·9Н2О). Под действием кислорода воздуха растворы их быстро окрашиваются в красноватый цвет вследствие образования аналогичных полисульфидам полиселенидов и полителлуридов. Известны, в частности, Nа2Se6 и Nа2Те6, но производные Sе и Те типа многосернистых водородов не получены. Селениды и теллуриды большинства металлов в воде нерастворимы.
Возможность образования Н2Ро была установлена по радиоактивности газа выделяющегося при обработке соляной кислотой магния, на котором был перед тем осажден полоний. Гидрид полония еще менее стоек, чем Н2Те, из-за чего и не мог быть выделен, но производящиеся от него полониды (Nа2Ро и др.) известны. Теплота образования РоH2 из элементов оценивается в — 188 кДж/моль.
Все галоидные соединения селена и теллура могут быть получены путем взаимодействия элементов. Известны следующие галогениды:
| Состав | SeF6 | SeF4 | SeCl4 | Se2Cl2 | SeBr4 | Se2Br2 |
| Агрегатное состояние | газ | жидк. | тверд. | жидк. | тверд. | жидк. |
| Цвет | бесцв. | бесцв. | бесцв. | коричн. | желт. | красн. |
| Состав | ТеF6 | TeF4 | TeCl4 | TeCl2 | TeBr4 | TeBr2 | TeI4 |
| Агрегатное состояние | газ | тверд. | тверд. | тверд. | тверд. | тверд. | тверд. |
| Цвет | бесцв. | бесцв. | бесцв. | зелен. | оранж. | коричн. | серо-черн. |
Молекулы галогенидов ЭF6 имеют структуры октаэдров с атомом Э в центре [в(SеF) = 169 пм, d(ТеF) = 182 пм]. Подобно SF6, гексафторид селена и теллура характеризуются высоким давлением паров в твердом состоянии (т. возг. соответственно -46 и -39 °С). Поэтому их точки плавления ( -35 и -37 °С) могут быть определены лишь под повышенным давлением. Образование обоих соединений из элементов сопровождается значительным выделением тепла (1028 и 1317 кДж/моль).
По своему общему характеру галогениды селена похожи на соответствующие производные серы, причем тип Э2Г2 в данном случае менее, а тип ЭГ4 — более устойчив. Например, Sе2Сl2 даже при осторожном нагревании распадается на Sе и SеСl4, а последний, хотя и возгоняется с разложением на SеСl2 и Сl2, но вновь образуется из них при охлаждении. По строению молекулы SеF4 подобен SF4 с параметрами d(SеF1) = 168, d(SеF2) = 177 пм, РF1SеF1 = 100°, РF2SеГ2 = 169°. Для SеF4 (т. пл. -10, т. кип. 108 °С), SеСl4 (теплота образования из элементов 188 кДж/мюль, т. возг. 196 °С) и SеВr4 известны двойные соединения с галогенидами (главным образом типа SеГ4·2МГ) и серным ангидридом (например, бесцветный SеСl4·SО3). Описано также соединение состава НgSеF4, образующееся при взаимодействии SеF4 с ртутью. Водой почти все галогениды селена легко разлагаются. Наиболее медленно протекает гидролиз SеF6.
Интересен жидкий при обычных условиях смешанный хлорид серы и селена типа Э2Сl2, который может быть, по-видимому, получен в двух формах — темно-красной (исходя из Sе и S2Сl2) и бледно-оранжевой (исходя из S и Sе2Сl2). Различие обеих форм естественно объяснялось бы их разной структурой: по типу SеSСl2 и SSеСl2. Такая трактовка, предполагающая четырехвалентность центрального атома не соответствует структуре S2Cl2, но находится в хорошем согласии со строением более устойчивой формы S2F2.
Галогениды теллура уже резко отличаются по свойствам от соответствующих производных серы. Тогда как четырехфтористая сера при обычных условиях газообразна, ТеF4 плавится лишь при 130 °С. В отличие от шестифтористой серы ТеF6 довольно легко разлагается водой и способен давать продукты присоединения (известен ТеF6·2СsF). С другой стороны, иодиды серы (и селена) вообще не получены, а черный ТеI4, образуется при совместном растирании элементов в присутствии воды. Теплоты образования ТеСl4 и ТеВr4 из элементов равны соответственно 322 и 196 кДж/моль. Тип Те2Г2 для теллура неизвестен, соединения же, отвечающие типам ТеГ2 и ТеГ4, имеют скорее характер не галогенангидридов, а солей. Водой они разлагаются лишь частично, причем для типа ТеГ2 наряду с гидролизом наблюдается распад по схеме: 2 ТеГ2 = ТеГ4 + Те. Определение пространственного строения ТеВr2 показало, что молекула треугольна [d(ТеВr) = 251 пм, Рa = 98°].
Некоторые из рассматриваемых соединений не разлагаются ни при своих довольно высоко лежащих точках плавления (ТеСl4 — 224 °С, ТеСl2 — 208 °С, ТеВr4 — 380 °С, ТеВr2 —280 °С), ни при температурах кипения (ТеСl4 — 420 °С, ТеСl2 — 328 °С, ТеВr2 — 340 °C). Напротив, ТеI4 начинает разлагаться уже выше 100 °С.
Для галогенидов типа ТеГ4 характерно образование продуктов присоединения с соответствующими галоидоводородными кислотами и особенно некоторыми их солями. Наиболее обычные из них отвечают общей формуле вида М2ТеГ6, где М — одновалентный металл [d(ТеСl) = 254, d(ТеВr) = 270 пм]. Хлориды имеют желтую окраску, бромиды — оранжевую и иодиды — черную. Для фторидов характерен другой тип — МТеF5 (где М — Сs, Rb, К). Водой эти соли тотчас разлагаются.
Для полония были получены (в миллиграммовых количествах) следующие галогениды:
РoСl4 РoВr4 РoI4 РoСl2 РоВr2
желтый красный черный красный коричневый
Все они представляют собой твердые вещества.
Тетрахлорид плавится около 300 °С и кипит при 390 °С. Известны также двойные соединения типов РоГ4·МГ и РоГ4·2МГ. Для расстояний Ро-Сl и Ро-Вr (в производных второго типа) были найдены значения 238 и 261 пм.
При нагревании в токе воздуха или кислорода селен и теллур сгорают с образованием диоксидов. Обе они представляют собой бесцветные кристаллические вещества, сильно отличающиеся друг от друга по растворимости в воде: у SeO2 она весьма велика, у TeO2 — очень мала.
При получении SеО2 сжиганием селена (сгорающего синим пламенем) воздух или кислород полезно предварительно насытить окислами азота (пропуская его сквозь дымящую НNО3), так как сгорание идет в этом случае гораздо быстрее. Теплота образования диоксида селена из элементов равна 226 кДж/моль, а средняя энергия связи Sе=О оценивается в 426 кДж/моль. Кристаллический селендиоксид образован неплоскими цепями -O-Sе(O)O-Sе(O)- с параметрами d(OSе) = 178, d(SеО) = 173 пм, РOSеО = 98°, РSеOSе = 125° и при нагревании возгоняется (т. возг. 337 °С, теплота возгонки 92 кДж/моль). Желтовато-зеленый пар SеО2 имеет характерный запах (“гнилой редьки”) и слагается из отдельных молекул [d(SеО) = 161 пм]. Сухой диоксид селена легко образует продукты присоединения. Примером может служить жидкий при обычных условиях (и устойчивый до 170 °С, когда он перегоняется с частичным разложением) желтый SеО2·2НС1.
Получение ТеО2 (т. пл. 733, т. кип. 1257 °С) удобнее вести не сжиганием теллура (сгорающего зеленовато-синим пламенем), а окислением его крепкой НNО3. При упаривании или разбавлении водой полученного раствора диоксид теллура осаждается из него в виде бесцветных кристаллов, при нагревании желтеющих. Теплота образования ТеО2 из элементов составляет 322 кДж/моль, а средняя энергия связи Те=О оценивается в 247 кДж/моль. Растворимость теллурдиоксида в воде очень мала, но он растворим в растворах сильных щелочей и кислот (с образованием солей). Так, в 1 н. НСl может быть при 25 °С получен 0,01 М раствор ТеО2. Из характерных для диоксида теллура продуктов присоединения наиболее интересен устойчивый до 300 °С (но тотчас разлагаемый водой) 2ТеО2·НСlO4.
Подобно SO2, диоксиды селена и теллура являются кислотными ангидридами: при растворении их в воде образуются соответственно селенистая (H2SeO3) и теллуристая (H2TeO3) кислоты. Обе они диссоциированы несколько слабее сернистой.
Соли селенистой кислоты (селенистокислые, или селениты) могут быть получены нейтрализацией растворов H2SeO3, соли теллуристой (теллуристокислые, или теллуриты) — растворением TeO2 в щелочах. И селениты, и теллуриты, как правило, бесцветны. По растворимости селениты в общем сходны с соответствующими сульфитами, тогда как из теллуритов хорошо растворимы в воде только производные наиболее активных одновалентных металлов (Na, K и др.).
В то время как для четырёхвалентной серы восстановительные свойства характернее окислительных, для SeIV и TeIV имеет место обратное: они довольно легко восстанавливаются до элементарных Se и Te. Напротив, перевод четырёхвалентных селена и теллура в шестивалентное состояние может быть осуществлён лишь действием наиболее сильных окислителей.
Свободная селенистая кислота (К1 = 2·10-3, K2= 5·10-9)может быть получена растворением порошкообразного селена в разбавленной НNО3 по реакции:
3 Sе + 4 НNО3 + Н2О = 3 Н2SеО3 + 4 NО
При упаривании раствора она выделяется в виде бесцветного кристаллогидрата, расплывающегося во влажном воздухе и постепенно выветривающегося в сухом. Молекула Н2SеО3 существует только в форме SеО(ОН)2. Она устойчива лишь ниже 70 °С, а выше этой температуры распадается на SеО2 и воду даже в растворе. По другим данным, первичным продуктом термического разложения является Н2Sе2О5.
Окислительные свойства селенистой кислоты выражены не особенно сильно она окисляет SO2 и I-, но не способна окислить Вr-. Ее взаимодействие с КMnO4 протекает по уравнению:
15 Н2SеО3 + 10 КМnO4 = 5 К2SеO4 + 10 Н2SеО4 + 10 МnО2 + 5 Н2О
Из солей Н2SеО3 следует отметить труднорастворимый селенит серебра Аg2SeO3 (ПР = 1·10-15). Ион SеО32- представляет собой трехгранную пирамиду с параметрами: d(SеО) = 169 пм и РOSеО = 101°.
Наличие у селенистой кислоты очень слабо выраженной основной функции более отчетливо выявляется при ее взаимодействии с безводной НСO4. Реакция в этих условиях идет по уравнению:
SеО(ОН)2 + НСlO4 = [Sе(ОН)3]СlO4
Образующийся солеобразный продукт содержит пирамидальный катион [Sе(ОН)3]+ и представляет собой бесцветное и очень гигроскопичное кристаллическое вещество (т. пл. 33 °С). Аналогичными свойствами обладает сульфат селенила — (SеО)SO4, который может быть получен взаимодействием SеО2 и SO3 при нагревании (в запаянной трубке). Известен фторсульфонат селенила — SеО(SОзF)2.
Теллуристая кислота получена в виде кристаллогидрата Н2ТеO3·Н2О. Она обладает свойствами амфотерного электролита. Ее кислотная функция (К1 = 2·10-3, К2 = 2·10-8) выражена значительно сильнее основной (К1 = 3·10-11). Последняя проявляется при растворении ТеО2 в концентрированных сильных кислотах — происходит образование солей четырехвалентного теллура, например, по схеме:
ТеО2 + 4 НI = ТеI4 + 2 Н2О
Помимо галогенидов, в твердом состоянии были получены также основные сульфаты и нитраты четырехвалентного теллура.
Окислительные свойства теллуристой кислоты выражены несколько слабее, чем у селенистой. Анион ТеО32- имеет строение треугольной пирамиды с параметрами d(ТеО) = 188 пм, РОТеО) = 100°.
И для селенистой и особенно для теллуристой кислот весьма характерно образование солей типа М2О·nЭО2, причем для селена известны производные с n = 2 и 4, а для теллура — с n = 2, 4 и 6. Аналогичными им соединениями серы являются соли пиросернистой кислоты, в которых n = 2.
Из других производных рассматриваемых элементов в их четырехвалентном состоянии следует отметить оксогалогениды селена. Очень ядовитый SеОСl2 образуется при совместном нагревании SеСl4 и SеО2 и представляет собой желтоватую жидкость (т. пл. 11, т. кип. 178 °С). Он является хорошим растворителем многих веществ, в частности растворяет серу, селен, теллур, бром и иод. Смеси хлористого селенила с SО3 способны растворять многие окcиды металлов (Сr2О3, Аl2O3 и т. д.). Для SеОСl2 довольно характерны окислительные свойства (особенно при нагревании). Водой он разлагается на НСl и селенистую кислоту.
Известны также бесцветный SеОF2 (т. пл. 5, т. кип. 125 °С) и желтый SеОВr2 (т. пл. 42, т. кип. 217 °С с разложением).
Диоксид полония (РоО2) образуется из элементов при 250 °С в виде красных кристаллов (т. возг. 885 °С), постепенно переходящих при хранении в более устойчивую при обычных условиях желтую форму. С химической стороны она аналогична диоксиду теллура, но отвечающая ей гидроксид обладает более основными свойствами. Так, для теллура известны лишь основные соли кислородных кислот, тогда как для полония были получены и средние соли (в частности, бесцветные Ро(SО4)2 и Ро(NО3)4]. Напротив, образующиеся при взаимодействии РоО2 или его гидроксида с сильными щелочами соли полонистой кислоты (Н2РоО3) — полониты — гидролизованы значительно сильнее теллуритов ([К2РоО3]/[КОН]2 = 8·10-5).
Такое окисление идет, например, по схеме:
5 H2ЭO3 + 2 HClO3 = 5 H2ЭO4 + Cl2 + H2O.
Образующиеся селеновая (H2SeO4) и теллуровая (H2TeO4) кислоты представляют собой бесцветные кристаллические вещества, хорошо растворимые в воде. Селеновая кислота по силе близка к серной, тогда как теллуровая является кислотой весьма слабой.
Соли селеновой кислоты (селеновокислые, или селенаты) по свойствам похожи на соответствующие сульфаты. Напротив, соли теллуровой кислоты (теллуровокислые, или теллураты) существенно отличаются от них. Например, BaTeO4 выделяется из раствора с кристаллизационной водой и легко растворяется в соляной кислоте. В воде хорошо растворимы лишь теллураты наиболее активных одновалентных металлов.
Обе рассматриваемые кислоты являются сильными окислителями и, например, с HCl взаимодействуют по схеме:
H2ЭO4 + 2 HCl Ы H2ЭO3 + Cl2 + H2O.
В кислой среде равновесие смещается вправо, в щелочной — влево.
И для селеновой, и для теллуровой кислот характерна медленность проявления их окислительного действия (особенно — в разбавленных растворах). В ряде случаев окислительно-восстановительный процесс из-за этого практически не протекает. Относительно быстрее других восстановителей окисляются обеими кислотами галоидные ионы (I’, Вr’, Сl’). Селеновая кислота является более сильным окислителем, чем теллуровая.
Свободная селеновая кислота (К2 = 1·10-2) проще всего получается обработкой взвеси Аg2SеО3 бромной водой. Реакция идет по уравнению:
Аg2SеО3 + Вr2 + Н2О = 2 АgВrЇ + Н2SеO4
Осадок АgВr отфильтровывают, а избыток бромида удаляют кипячением жидкости. Упариванием последней в вакууме и кристаллизацией остатка селеновая кислота может быть выделена как в виде кристаллогидрата Н2SеO4·Н2О (т. пл. 26 °С), так и в безводном состоянии (т. пл. 62 °С). Известны также кристаллогидраты селеновой кислоты с 2Н2О (т. пл. -24 °С), 4Н2О (т. пл. -52 °С) и 6Н2О (т. пл. -68 °С).
В расплавленной безводной селеновой кислоте имеет место равновесие по схеме:
2 Н2SеO4Ы Н3О++ НSе2О7
При нагревании она растворяет не только серебро (как Н2SO4), но и золото. Однако платина в ней не растворяется. Выше 260 °С селеновая кислота переходит в SеО2. Подобно серной, она очень гигроскопична и обугливает многие органические вещества. Сила селеновой кислоты оценивается в 0,9 от серной. Ион SеO42- имеет структуру тетраэдра с расстоянием d(SеО) = 165 пм.
Соли селеновой кислоты легко образуются при действии хлора на щелочные растворы селенитов или сплавлении селенитов с КNО3. Из реакционной смеси обычно выделяют малорастворимый (около 2·10-4 моль/л при обычных условиях) ВаSеО4, обменным разложением которого с сульфатами других металлов можно получать их селенаты. Последние в общем похожи на соответствующие сульфаты, но лучше растворимы в воде и менее устойчивы по отношению к нагреванию.
Производным селеновой кислоты является также имеющий суммарную формулу Sе2О5 селенат селенила — (SеО)SеO4, по способу образования и свойствам аналогичный его сульфату. Удобнее получать это бесцветное кристаллическое вещество выдерживанием расплавленного SеО3 при 170 °С или взаимодействием SеО3 с SеО2 в жидкой SO2. В высоком вакууме он при 145 °С возгоняется без разложения, а выше 185 °С переходит в SеО2.
Свободная теллуровая кислота (К1 = 2·10-8, К2 = 1·10-11, К3 = 3·10-15) может быть получена взаимодействием элементарного теллура с 30 %-ной Н2О2 (при нагревании на водяной бане). Выделяется она в виде бесцветного кристаллогидрата Н2ТеО4·2Н2О. Как и у иодной кислоты водороды воды, входящей в состав этого кристаллогидрата, способны частично или полностью замещаться на металл. Например, известны соли состава Аg6ТеО6 и Нg3ТеО6, отвечающие шестиосновной ортотеллуровой кислоте — Н6ТеО6.
Хотя нагреванием последней при 100ё200 °С может быть получен порошок состава Н2ТеО4, однако эта форма для теллура нехарактерна: все двузамещенные теллураты производятся от ортотеллуровой кислоты. При температурах ниже 10 °С сама она выделяется из раствора в виде кристаллогидрата H6TeO6··4H2O. Структура Н6ТеО6 отвечает правильному октаэдру с теллуром в центре и гидроксильными группами в вершинах. В концентрированных растворах она несколько полимеризована.
Соли теллуровой кислоты удобно получать сплавлением теллуритов с КNО3 (нормальные соли состава М2ТеО3 при 450 °С окисляются уже кислородом воздуха).
Наиболее обычными теллуратами являются довольно малорастворимый Nа2Н4ТеО6 и легкорастворимый К2Н4ТеО6·3Н2О. Шестизамещенная соль натрия (Nа6ТеО6) может быть получена сплавлением Н6ТеО6 с NаОН. На воздухе она постепенно переходит в Nа2H4ТеО6·3Н2О. По данным рентгеноструктурного анализа Нg3ТеО6, ион ТеО66- имеет структуру октаэдра с d(ТеО) = 198 пм.
При нагревании Н6ТеО6 в запаянной трубке до 140 °С образуется аллотеллуровая кислота — вязкая жидкость, полностью смешивающаяся с водой и обладающая ясно выраженными кислотными свойствами. Она представляет собой, по-видимому, раствор смеси полимерных теллуровых кислот. При хранении водного раствора аллотеллуровая кислота постепенно переходит обратно в Н6ТеО6.
Отвечающий селеновой кислоте ангидрид — триоксид селена SeO3 представляет собой бесцветное кристаллическое вещество, хорошо растворимое в воде с образованием селеновой кислоты. Напротив, жёлтый триоксид теллура (TeO3) в воде почти нерастворим. Однако концентрированные растворы сильных щелочей растворяют его с образованием соответствующих теллуратов.
Триоксид селена (т. пл. 121 °С) удобно получать по схеме:
К2SеО4 + SO3 = К2SO4 + SеО3
Теплота его образования из элементов равна 171 кДж/моль. Основной молекулярной формой этого вещества для всех агрегатных состояний является тетрамер (SеО3)4 (рис. 4), в котором атомы селена [d(SеSе) = 313 пм соединены друг с другом кислородными мостиками (d(SеО) = 180 пм] и имеют по два “собственных” атома кислорода [d(SеО) = 156 пм]. В парах присутствуют и мономерные молекулы SеО3 (при 120 °C порядка 25 %) с d(SеО) = 169 пм. Расплавленный селентриоксид весьма склонен к переохлаждению, а твердый известен в двух формах (из которых одна нестабильна). Выше 180 °С он разлагается на SеО2 и кислород, но в вакууме может быть возогнан без разложения.
Рис. 4. Схема строения (SeO3)4.
Cелентриоксид обладает настолько сильными окислительными свойствами, что окиcляет НСl до свободного хлора даже при охлаждении. Однако из раствора в жидкой SO2 он может быть выделен без изменения. Избытком серы SеО3 восстанавливается до элементарного селена, а при взаимодействии его с селеном или теллуром образуются зеленые вещества, по-видимому, аналогичные S2O3.
При растворении SеО3 в Н2SеO4 образуются более или менее нестойкие пирокислоты селена — Н2Sе2О7 (т. пл. 19 °С), Н4Sе3О11 (т. пл. 25 °С с разл.).
Триоксид теллура может быть получена обезвоживанием теллуровой кислоты при 300-350 °С. Образующаяся при этом желтая ТеО3 аморфна и крайне малорастворима в холодной воде (примерно 0,5 г/л). Соляную кислоту ТеО3 окисляет лишь при нагревании (в отличие от SеО3).
Длительным выдерживанием ТеО3 при 406 °С был получен светло-желтый оксид состава Те2О5, переходящий в ТеО2 лишь при 485 °С. Он плохо растворим в воде но хорошо растворяется в крепком растворе КОН. Данные магнитного исследования говорят за то, что этот оксид является производным пятивалентного теллура.
56
Четвертая группа периодической системы.
По электронным структурам нейтральных атомов к углероду и кремнию примыкают германий и его аналоги. Максимальная валентность этих элементов, как по отдаче, так и по присоединению электронов, равна четырем. В связи с увеличением объема атомов при переходе от углерода к свинцу процесс принятия электронов ослабевает, а лёгкость их потери возрастает, поэтому металлические свойства атомов возрастают сверху вниз.
Из-за наличия во внешнем слое атомов лишь двух электронов, у титана и его аналогов отсутствует тенденция к дополнению внешнего слоя до октета. Вместе с тем в положительной валентности будет наблюдаться сходство подгруппы титана с кремнием.
УГЛЕРОД
Кларк углерода 0,14%, но тем не менее значение углерода исключительно велико, так как его соединения являются основой всех живых организмов.
Формы нахождения углерода в природе многообразны. Кроме тканей живых организмов и продуктов их разрушения (каменный уголь, нефть и т. д.), он входит в состав многих минералов, имеющих большей частью общую формулу МСО3, где М – двухвалентный металл. Наиболее распространенным из таких минералов является кальцит (CaCO3), образующий иногда громадные скопления на отдельных участках земной поверхности. В атмосфере углерод содержится в виде углекислого газа, который в растворенном состоянии находится также во всех природных водах.
В форме древесного угля углерод был известен человечеству с незапамятных времен. Современное название он получил в 1787 г.
Природный углерод слагается из двух изотопов — 12С (98,892%) и 13С(1,108%). Масса изотопа углерода-12 принята за единицу атомных и молекулярных масс. В различных природных объектах соотношение обоих изотопов может незначительно изменяться. Поэтому атомный вес углерода дается с точностью ± 5•10-5.
Свободный углерод встречается в природе в виде двух простых веществ — алмаза и графита. К ним можно отнести и так называемый “аморфный” углерод, простейшим представителем которого является древесный уголь. Алмаз имеет плотность 3,5 г/см3 и является самым твёрдым из всех минералов. Наиболее чистые алмазы бесцветны и прозрачны. Графит представляет собой серую, имеющую металлический блеск и жирную на ощупь массу с плотностью 2,2 г/см3. Он очень мягок — легко царапается ногтем и при трении оставляет серые полосы на бумаге. “Аморфный” углерод по свойствам довольно близок к графиту. Плотность его колеблется в пределах 1,8-2,1 г/см3. У некоторых разновидностей “аморфного” углерода очень сильно выражена способность к адсорбции (т.е. поглощению на поверхности) газов, паров и растворённых веществ.
Тройной точке на диаграмме состояния углерода отвечает температура около 3700 °С и давление около 110 атм. Поэтому при нагревании под обычным давлением (в отсутствие воздуха) углерод не плавится, а возгоняется.
Наиболее устойчивой формой углерода при обычных условиях является графит. Теплота его сгорания (до СО2) 393 кДж/моль. У алмаза она 395 кДж/моль, а у “аморфного” углерода 400-410 кДж/моль. Переход менее устойчивых форм в графит при обычных условиях не происходит, но выше 1500 °С (в отсутствие воздуха) он идёт довольно быстро.
Теплота плавления графита (при 47 тыс. атм) составляет 105 кДж/моль. При плавлении разрывается только часть связей кристаллической решетки.
Давление пара графита даже при 2500 °С ничтожно мало (примерно 5·10-6 атм), и температура его возгонки около 3700 °С. Пары углерода состоят из отдельных атомов и более сложных образований общей формулы Сп. При 3100 °С пар состоит в основном из молекул С=С (131 пм) с энергией диссоциации 600 кДж/моль, а при дальнейшем повышении температуры он, по-видимому, обогащается молекулами С=С=С, С=С=С=С и т. д.
Образование природных алмазов происходило путем кристаллизации углерода в глубинных слоях Земли (200-300 км от поверхности) при температурах порядка 3000 °С и давлениях порядка 200 тыс. атм. Их коренные месторождения связаны с весьма редким выходом на поверхность особой горной породы — кимберлита, а рассыпные изредка встречаются в наносных пластах. Промышленные разработки содержат в среднем только 0,5 г алмаза на тонну породы. Богатые месторождения были открыты в Якутии (1955 г).
Рис. 1. Схема расположения атомов С в алмазе. Рис. 2. Обычная огранка бриллианта.
Структуру алмаза можно представить в виде тетраэдров с атомом углерода в центре, которые повторяются в бесконечности в трех измерениях (рис. 1). Алмаз имеет атомную кристаллическую решетку.
Несмотря на свою твердость, алмаз хрупок и легко раскалывается от удара. Он хорошо проводит тепло, но практически не проводит электрический ток. Не все алмазы бесцветны, некоторые из них имедт окраску, от лишь слегка наметившейся до интенсивной. По отношению к рентгеновским лучам алмаз прозрачен (в отличие от подделок), а для ультрафиолетовых одни кристаллы прозрачны, другие нет.
Алмаз отличается большой инертностью: на него не действуют ни кислоты, ни щелочи. На воздухе алмаз горит при температере около 900°С, а в кислороде — около 700 °С. После сгорания остаётся немного золы (0,02 вес. % и более), что свидетельствует о наличии в природных алмазах примесей (главным образом алюминия, кремния, кальция и магния). При нагревании выше 1200 °С в отсутствие воздуха начинается графитизация алмаза.
В кристаллах алмазов обычного типа небольшая (порядка 1:1000) часть атомов углерода заменена на атомы азота. Из представителей более редкого типа особенно интересны светло-голубые алмазы, электропроводность которых сравнительно выше, чем у прочих образцов. При нагревании выше 100 °Сони приобретают полупроводниковые свойства р-типа, сохраняющиеся в атмосфере водорода до 1100 °С.
Наиболее красивые алмазы шлифуют и под названием бриллиантов (рис. 2) употребляют в качестве украшений. Для их расценки служит применяемая к драгоценным камням единица массы — карат (0,2 г). Самый крупный добытый алмаз (“Куллинан”) весил 3026 каратов, т.е. более 600 г.
Исключительная твердость алмаза обусловливает его ценность для техники. Промышленность использует все те камни (громадное большинство), в которых имеется какой-либо изъян (некрасивая окраска, трещины и т. д.), делающий их непригодными в качестве украшений.
Сравнительно невысокая цена таких камней с браком позволяет употреблять их непосредственно или в форме алмазного порошка для заточки и шлифовки режущих инструментов из твердых сплавов, правки шлифовальных кругов, при буровых работах в горном деле (алмазное бурение), резке стекла и твердых каменных пород, сверлении стали, обточке металлических валов. Использование алмазов резко повышает скорость и качество обработки самых разнообразных материалов.
“Если бы США были отрезаны от их современных источников алмазов, то их промышленный потенциал за очень короткий срок упал бы наполовину.” (Левис). Ежегодная мировая добыча алмазов около 5 т.
Существует предположение, что исходным материалом для природного синтеза алмазов служил углерод, возникавший в результате восстановления (при высоких температурах и под большим давлением) карбонатных пород двухвалентным железом по примерной суммарной схеме:
СаСО3 + 5 FeO = Ca(FeO2)2 + Fe3O4 + C.
Необходимое для кристаллизации углерода в форме алмаза очень высокое давление создавалось за счет его случайных местных повышений.
Попытки искусственного получения алмазов предпринимались многократно, но впервые увенчались успехом лишь в 1953 г. Перевод графита в алмаз может быть осуществлен только при очень высоких давлениях, при высоких температурах и наличии катализаторов, из которых наиболее подходящими оказались некоторые элементы триад. Зародышевые кристаллы алмаза возникают на поверхности раздела между графитом и расплавленным металлом-катализатором. Они остаются покрытыми пленкой жидкого углеродсодержащего металла, сквозь который углерод затем и диффундирует от графита к алмазу по мере его роста. Современная техника позволяет получить в одной камере за несколько минут 20 г алмазов.
Интересен также другой метод синтеза — действием на графит (в смеси с катализатором) ударной волны, создаваемой взрывом. Мгновенность этого действия компенсируется возникновением в момент взрыва чрезвычайно высоких давления и температуры. Так при одном из опытов с ударной волной под давлением в 300 тыс. атм. почти весь взятый графит превратился в очень мелкие алмазные кристаллики (размером до 40 мк).
Искусственные алмазы представляют собой мелкие кристаллы, преимущественная форма которых обычно меняется от кубической (при сравнительно низких температурах синтеза) к октаэдрической (при высоких). Цвет их тоже различен: от черного при низких температурах до зеленого, желтого и белого — при высоких. Например, в одном из опытов под давлением 200 тыс. атм. мгновенным (в течение тысячных долей секунды) нагреванием графита электрическим разрядом до 5000 °С были получены бесцветные алмазы чистой воды. Цвет искусственных алмазов существенно зависит от природы включаемых в кристаллы примесей (а тем самым и от состава исходной графитовой смеси). Например, примесь никеля придаёт зеленоватые тона, а одновременно никеля и бора — синие.
Интересны полупроводниковые алмазы (р-типа), синтезированные при 1100 °С и 70 тыс. атм. в присутствии небольших добавок В, Ве или А1. На основе их хорошей теплопроводности и сильной зависимости электрического сопротивления от температуры был сконструирован миниатюрный и чувствительный термометр (алмазный термометр) с областью применения от -200 до +650 °С.
Из-за своих малых размеров и обычно некрасивой окраски синтетические алмазы в качестве украшений почти не используются. Напротив, по техническим качествам они лучше естественных. Ежегодное мировое производство алмазов по общему объёму производства соизмеримо с добычей.
Путём выращивания затравочных кристаллов алмазы могут быть синтезированы и вне области их устойчивости. Медленным пропусканием метана под давлением 0,01 атм над нагретым до 1100 °С зародышевым кристаллом достигалась скорость их роста до 0,5% в час. Процесс сводится к термическому разложению метана, причём освобождающийся атомарный углерод осаждается на поверхности зародышевых кристаллов, продолжая их структуру. Подобным путём были, в частности, получены нитевидные кристаллы (“усы”) алмаза длиной до 2 мм (при диаметре в несколько десятком микрон).
Кристалл графита построен из плоских углеродных атомов, располагающихся точно друг над другом через одну, т.е. с чередованием по типу АБАБ… Известна также гораздо более редкая (и менее устойчивая) АБВАБВ...
Каждый атом углерода в плоскости сетки (“паркета”) соединён ковалентными связями с тремя другими. Связи эти значительно короче (147 пм), чем в алмазе, что указывает на их высокую прочность. Расстояние между отдельными слоями велико (335 пм), и связь между ними слаба (17 кДж/моль). Внешне это выражается в лёгкой расщепляемости графита по плоскостям спаянности кристалла на отдельные тонкие пласты (“чешуйки”).
Графит хорошо проводит тепло (в 3 раза лучше ртути) и обладает близкой к металлам электропроводностью; больше параллельно слоям, чем перпендикулярно им. Максимум теплопроводности графита наблюдается при 0 °С, а электропроводности — около 600 °С. Механическая прочность графита при переходе от обычных температур к 2500 °С возрастает почти вдвое. Его сжимаемость примерно в 20 раз больше сжимаемости алмаза. Заметное окисление графита при нагревании на воздухе наступает лишь выше 700 °С.
Относительно электронного строения графита имеются две основные точки зрения. Согласно одной из них, четвёртый валентный электрон каждого атома углерода участвует в формировании связей внутри сетки (повышая их порядок до 1,33), а связь между слоями осуществляется лишь межмолекулярными силами. Согласно другой точке зрения, четвёртые валентные электроны атомов углерода образуют слабые металлические связи между слоями (чем и обусловлены черты сходства графита с металлами). Вероятнее всего, наиболее правильно сочетание обеих трактовок с преобладанием первой из них. Экспериментально было установлено, что свободные электроны в графите имеются, но эффективное их число сравнительно мало — около 6·1018 на 1 см3 (т.е. один электрон приходится примерно на 18 тыс. атомов углерода).
Интересной особенностью графита является его способность поглощать значительные количества некоторых веществ за счет их внедрения в пространства между молекулярными слоями. Подобно другим аддуктам, соединения графита характеризуются в общем переменными составами, которые ограничиваются некоторыми предельными. Как правило, последние не отвечают валентным соотношениям, характерным для углерода и соответствующих элементов. Образование всех продуктов внедрения сопровождается существенным увеличением расстояния между углеродными сетками, но при помощи подходящих воздействий поглощенные вещества могут быть извлечены с более или менее полным восстановлением исходной структуры “хозяина”.
Все графитиды целесообразно разделить на две группы. К первой можно отнести только производные фтора и кислорода, характеризующиеся возникновением ковалентных связей между атомами углерода и внедряющихся элементов, что ведёт к большему или меньшему искажению структуры “паркетов”. Относящиеся сюда вещества предельных составов по своему характеру близки к истинным химическим соединениям.
Вторая группа охватывает все остальные многочисленные продукты внедрения в графит. Они близки к типичным аддуктам, от которых отличаются главным образом возникновением некоторой разноимённой поляризации внедрённых частиц в графитных “паркетах”. Последние при этом не подвергаются сколько-нибудь существенному искажению.
Наиболее чётко выраженный характер истинного химического соединения имеет фтористое производное графита. Взаимодействие последнего со фтором при 450 °С ведёт к медленному образованию продуктов внедрения состава СFп, где п і 1. Обычно получаются чёрные или серые фториды с n > 1, но иногда удаётся получить предельный продукт состава CF. Его образование протекает с возникновением ковалентных связей C-F, увеличением расстояния между слоями графита до 660 пм и изменением самой структуры этих слоёв от плоской к складчатой с расстоянием С-С 154 пм. Интересно, что меньшему содержанию фтора отвечает большее расстояние между слоями (до 880 пм для СF0,68). Фторид представляет собой серебристо-белое, в тонких слоях прозрачное вещество, не проводящее электрический ток и чрезвычайно химически стойкое (не взаимодействует ни с кислотами, ни со щелочами). Однако длительным действием водорода в момент выделения (цинковая пыль + уксусная кислота) фтор может быть извлечён с восстановлением графитной структуры. Нагревание CF выше 500 °С сопровождается энергичным (вплоть до взрыва) разрушением этого вещества с образованием летучих фторидов углерода (CF4, C2F6 и др.) и выделением сажи.
Рис. 3. Схема структуры слоя CF. Рис. 4. Схемы структуры слоёв в продуктах окисления графита.
Действие на графит смеси фтора с избытком фтористого водорода при обычных условиях ведёт к увеличению расстояния между слоями до 540 пм и образованию чёрного вещества предельного состава С4F, также содержащего ковалентные связи С-F с расстоянием F-F 490 пм. Сами углеродистые “паркеты” при этом не изменяются, но становятся расположенными точно друг над другом (структура АААА...). Этот фторид проводит электрический ток, однако, примерно в 1000 раз хуже графита. Химически он очень устойчив, но при нагревании уже выше 100 °С начинает разлагаться.
Кислородсодержащие графитиды (“оксиды графита”, “графитовые кислоты”) образуются при длительном действии на графит сильных окислителей (например КС1О3 со смесью конц. серной и азотной кислот). После отмывки водой получают вещества коричневого, жёлтого или белого цвета. При сушке окисленного продукта над Р2О5 расстояние между слоями углеродных атомов 640 пм, при сушке на воздухе 900 пм, а в воде оно увеличивается до 1100 пм. Сами слои углеродных атомов графита претерпевают сильное искажение, предположительно за счёт возникновения связей С-О-С, С=О и С-О-Н.
Кислородные производные графита проявляют слабовыраженный кислотный характер. Они обладают также окислительными свойствами и под действием некоторых восстановителей (например НI и даже НBr) легко превращаются в графитоподобные продукты восстановления. В связи с наличием окислительных свойств предположили, что кислород в окисленном графите содержится в виде пероксидных групп (-О-О-), связывающих отдельные слои атомов углерода друг с другом. Это не противоречит и кислотным свойствам окисленного графита, происхождение которых может быть обусловлено гидролизом по схеме:
С-О-О-С + Н2О = СООН + СОН.
Вопрос о структуре окисленных форм графита нельзя считать окончательно разрешённым. При медленном нагревании этих форм происходит их возврат к структуре графита (с отщеплением СО2), а при быстром — распад с образованием СО, СО2 и сажи.
Для представителей второй группы продуктов внедрения в графит характерно наличие смещённых влево равновесий по схемам:
Сп + Х Ы Сп-+ Х+ .
Простейшими примерами таких систем могут служить производные калия и брома, предельные составы которых отвечают формулам С8К и С8Br.
Аддукт состава С8К образуется экзотермически (33 кДж/моль) при контакте графита с избытком жидкого или парообразного калия. Он имеет вид бронзы и обладает более высокой электропроводностью, чем исходный графит. Внедрение атомов калия не искажает “паркеты”, а вызывает их смещение в точно одинаковые позиции (структура АААА...). Расстояние от одного из них до другого становится при этом равным 540 пм, а каждый атом калия располагается между центрами двух шестиугольников, имея соседними 12 атомов С. Схема координации атомов в C8K показана на рис. 5. Аналогично калию ведут себя рубидий и цезий.
Рис. 5. Схема координации в C8K.
С химический точки зрения С8К характеризуется исключительной реакционной способностью. Он самовоспламеняется на воздухе и бурно — вплоть до взрыва — взаимодействует с водой, реагируя при этом как свободный щелочной металл (без образования каких-либо углеводородов). Металлическая ртуть извлекает калий с восстановлением структуры графита, а при действии на С8К жидкого аммиака происходит частичное замещение атомов калия молекулами NH3 по схеме:
3С8К + 4 NH3 = 2 C12K(NH3)2 + К.
Подобные же синие металло-аммиачные производные графита с расстоянием между “паркетами” около 660 пм могут быть получены прямым взаимодействием графита с раствором калия в жидком аммиаке. Вещества эти очень чувствительны к влаге, но на воздухе не самовоспламеняются.
Аддукт предельного состава С8Br образуется при взаимодействии графита с избытком жидкого брома или его паров (в последнем случае теплота образования около 33,5 кДж/моль). Он устойчив лишь при наличии избытка брома, тогда как в его отсутствие постепенно теряет почти весь бром.
Электропроводность этого аддукта значительно выше, чем у исходного графита. Расстояние между плоскостями “паркетов” возрастает при его образовании до 705 пм, причем промежуточные бромные слои образованы цепями из молекул Вr2 ( с ядерным расстоянием 213 и 224 пм). По всей вероятности, рассматриваемый аддукт наиболее правильно описывается равновесием С16 + Вr2 Ы С16+ + Вr2-(предполагалась также формула С56+Вr-•3Вr2). Значительно труднее брома внедряется в графит свободный хлор, тогда как иод вообще не внедряется. Вместе с тем IСl ведёт себя по отношению к графиту аналогично брому.
Графит выступает частичным донором электронов в “графитовых солях”. Они получаются действием на графит концентрированных кислот в присутствии сильного окислителя (или при анодном окислении). Например, синий бисульфат приблизительной формулы С24НSО4·Н2SО4. Известны также синие “соли” графита с анионами NO3-, ClO3-, HF2- и др. Все они характеризуются расстоянием между слоями углеродных атомов около 800 пм. При обработке соответствующей концентрированной кислотой каждая из этих “солей” обратимо превращается в другую. Действием воды или восстановителей (хлорид олова (II)) все они могут быть вновь переведены в графит.
К данной группе относятся и многочисленные продукты внедрения, возникающие при нагревании графита с безводными хлоридами ряда металлов (часто синтез лучше идёт в присутствии свободного хлора). Примером такого продукта внедрения может служить синее вещество приблизительного состава С8·АlCI3. В результате внедрения структура “паркетов” не изменяется, а расстояние между ними возрастает примерно до 950 пм. Водой некоторые из рассматриваемых веществ (например, Сn·AlCl3) разлагаются легко, тогда как некоторые другие (например, Cn·FeCl3) по отношению к ней весьма устойчивы.
Месторождения графита нередко обладают большой мощностью, оцениваемой миллионами тонн. Обычным исходным материалом для его образования служили останки растительности очень древних эпох. Лишь изредка встречаются месторождения, возникшие за счёт выделения углерода из расплавленных магм. Имеющий “минеральное” происхождение графит при сжигании почти не оставляет золы, тогда как обычно зольность его велика (от 1,5 до 15, а иногда даже до 35%). Ежегодная мировая добыча графита составляет 300 тыс. т.
Крупным потребителем графита является керамическая промышленность, изготовляющая из смеси графита с глиной тигли для переплавки металлов (графитовые тигли). Из прессованного графита делают газовые рули ракет. В металлургии он используется для обсыпки форм при литье. Из-за хорошей электропроводности графита из него изготовляют электроды для электротехнических и электрометаллургических процессов. Значительное количество графита идет для изготовления минеральных красок и (в смеси с глиной) карандашей. Интересным применением графита является использование его порошка (отдельно или вместе с машинным маслом) в качестве смазочного материала для трущихся частей механизмов.
Под вакуумом смазочные свойства графита исчезают, что связано с наличием при обычных условиях сорбированных между его слоями молекул газов воздуха.
Графит является основным (по объёму) конструкционным материалом большинства ядерных реакторов. Для этой цели он должен быть очень чист. Такой графит готовят искусственно, например длительным нагреванием (до 1500 и затем до 2800 °С) спрессованной смеси нефтяного кокса и каменноугольной смолы с последующим медленным охлаждением полученного продукта. В хороших искусственных графитах зольность не превышает тысячных долей процента. Интересной и практически важной разновидностью искусственного графита является пирографит, получаемый термическим разложением углеводородов на нагретой до 1000-2500°С поверхности. Его беспористый слой, повторяющий рельеф осадительной поверхности, характеризуется сильно выраженным различием свойств параллельно и перпендикулярно плоскостям отложения (например, параллельно им теплопроводность очень велика, а перпендикулярно очень мала). При температурах выше 2500 °С механическая прочность пирографита выше, чем у всех других известных материалов. Он начинает находить ряд важных применений в технике очень высоких температур (сопла ракет и др.), ядерной энергетике и химической промышленности.
При нагревании органических соединений до 500-800 °С в отсутствие воздуха происходит графитизация (т.е. сочетание углеродных атомов в подобные графиту структуры), причём форма частиц исходного вещества не изменяется. Процесс этот применяется главным образом для получения графитизированных волокнистых материалов, используемых затем в ряде областей техники.
Может существовать отличная и от графита, и от алмаза линейная форма элементарного углерода (карбин), слагающаяся из цепных полимеров типа (-СєС-СєС-)n — полиинов и (=С=С=)n —кумуленов. Исходя из ацетилена, был получен продукт, содержащий до 99,9% углерода и представляющий собой трёхфазную систему, в которой кристаллы полиина и кумулена сочетаются с аморфным углеродом. Он чёрного цвета, имеет плотность около 2,0 г/см3, ни в чём не растворяется, обладает свойствами полупроводника п-типа и переходит в графит выше 2000 °С. Теплота сгорания карбина — 356 кДж/моль — гораздо меньше, чем у других форм углерода.
Основными разновидностями “аморфного” углерода являются древесный уголь, животный уголь и сажа. Наиболее чистый “аморфный” углерод может быть получен обугливанием сахара.
Древесный уголь получают нагреванием древесины без доступа воздуха. Образующийся при этом рыхлый чёрный продукт сохраняет первоначальную структуру древесины. В металлургии им пользуются тогда, когда требуется особая чистота угля, например при рафинировании (очистке) меди. Ввиду большой адсорбционной способности древесного угля он применяется для очистки различных веществ от примесей и при изготовлении противогазов. Древесный уголь потребляется также при изготовлении чёрного пороха и в домашнем хозяйстве.
Животный уголь получают обугливанием животных остатков: костей (костяной уголь), крови (кровяной уголь) и т. д. Все виды животного угля характеризуются высокой адсорбционной способностью. Используется он главным образом в медицине (приём внутрь при некоторых отравлениях).
Сажа образуется при неполном сгорании многих органических соединений. Её частицы имеют сферическую форму со средним диаметром 10-300 нм. Обычно сажу получают, направляя пламя горящих с сильным выделением копоти веществ на охлаждаемую водой металлическую поверхность. Сажа широко используется резиновой промышленностью (ежегодное мировое потребление сажи 1,5 млн. т), так как входит в состав смесей для изготовления шин, калош и т. д. Сажа применяется и для изготовления красок (типографских, малярных, красок для кожи) и туши.
Кристаллическая структура этих видов “аморфного” углерода во всех исследованных случаях оказывалась тождественной структуре графита. Обычный “аморфный” углерод состоит в основном из очень мелких и беспорядочно расположенных кристаллов графита, однако в результате проводимой при 1000 °С реакции по схеме:
SiC + 2 Cl2 = SiCl4 + C
образуется углерод, не проявляющий никаких признаков кристаллической структуры, т.е. действительно аморфный.
Вместе с тем термическим разложением некоторых углеродистых материалов (синтетических смол и др.) может быть получен стекловидный углерод, также не имеющий определённой кристаллической структуры. Он состоит из графитоподобных микрослоёв, беспорядочно связанных друг с другом тетраэдрически координированными атомами углерода. Стекловидный углерод обладает рядом ценных свойств, в частности высокой устойчивостью к температурным, механическим и химическим воздействиям.
В обычных условиях углерод весьма инертен. Но при достаточно высоких температурах он становится химически активным по отношению к большинству металлов и многим неметаллам. “Аморфный” углерод значительно более реакционноспособен, чем графит и алмаз.
При нагревании “аморфного” углерода на воздухе он энергично взаимодействует с кислородом:
С + О2 = СО2 + 393 кДж.
В 1985 году была открыта совершенно новая форма углерода — фуллерит, принципиально отличающаяся и от графита, и от алмаза (и их модификаций). В противоположность двум последним, структура которых представляет собой периодическую решётку атомов, третья форма чистого углерода является чисто молекулярной. Отдельные частицы (фуллерены), из которых построено это вещество, представляют собой замкнутую поверхность сферы (или сфероида), составленную из атомов углерода. Сфера состоит из 20 правильных шестиугольников (подобных графитовым) и 12 правильных пятиугольников, образующихся при её формировании. Такая молекула С60 чрезвычайно напоминает футбольный мяч (d(С-С)=710 пм, диаметр сферы 1420 пм). Кроме того, были получены фуллерены С70, С74, С84 и т. п., имеющие форму сфероида (рис. 6).
Рис. 6. Молекулы С60 и С70.
В настоящее время фуллерены получают, пропуская электрический ток в вакууме через стержни из особо чистого графита. В месте контакта стержней возникает электрическая дуга, и графит начинает испаряться; на стенках камеры осаждается сажа. Затем камеру заполняют газообразным гелием, уносящим от отдельных фрагментов графита избыточную энергию. Из полученной смеси толуолом экстрагируют молекулы фуллеренов. Образующийся темно-красный раствор содержит молекулы С60 и С70 в пропорции 85:15. Дальнейшей хроматографической очисткой получают раствор чистого С60 (красный) или С70 (оранжевый). Упариванием такого раствора можно получить фуллерит — кристаллическое вещество с гранецентрированной кубической решёткой, образованной отдельными молекулами фуллеренов. При охлаждении до -24 °С решётка переходит в простую кубическую с увеличением объёма на 1 %. Интересно, что отдельные молекулы в кристалле фуллерита вращаются вокруг положения равновесия с частотой 1012 Гц. Плотность гексагонального фуллерита 1,7 г/см3, что в 1,35 раза меньше плотности графита. Известна также форма фуллерита, в которой молекулы расположены в вершинах тетраэдров, подобно атомам в кристалле алмаза.
Описанный выше способ получения фуллерита является чрезвычайно дорогостоящим, поэтому перспективной является непосредственная добыча фуллеренов из земных недр. Недавно было обнаружено, что минерал шунгит, огромные запасы которого находятся в Карелии, содержит до 0,1 % фуллеренов. К сожалению, еще не разработан промышленный метод выделения последних из шунгита.
Чистый фуллерит является полупроводником с шириной запрещенной зоны 1,5 эВ. Как он сам, так и его смеси с другими веществами (например, органическими полимерами) обладают фотопроводимостью: при облучении их даже видимым светом электрическое сопротивление резко падает. Фуллерит с добавками щелочных металлов при низких температурах (до 42,5 К для сплава RbTlC60) становится сверхпроводником.
Фуллерит не отличается химической активностью. В инертной атмосфере он стабилен до температуры 1200 К, в присутствии кислорода заметно окисляется уже при 500 К или (в обычных условиях) при облучении видимым светом. При этом образуются СО и СО2, а также аморфная структура с соотношением атомов С и О 5:1. Фуллерит хорошо растворим во многих неполярных растворителях (бензол, сероуглерод, толуол и т. п.).
Наиболее интересными из соединений фуллеренов являются продукты внедрения атомов различных элементов внутрь самой молекулы C60 — так называемые эндоэдральные соединения. В настоящее время более трети элементов периодической таблицы могут быть помещены внутрь молекулы фуллерена. В частности, имеются сообщения о получении подобных соединений для натрия, калия, рубидия, цезия, лантана, никеля. Предполагают, что молекулы фуллеренов с внедренными атомами редкоземельных элементов (тербий, гадолиний, диспрозий) будут становиться магнитными диполями, что позволяет, используя тонкие пленки таких соединений, создавать запоминающие устройства с громадной (4·1012 бит/см2) емкостью памяти.
Был получен полимер C60, в котором молекулы фуллеренов связаны бензольными кольцами; синтезированы также металлорганические полимеры (C60Pd)n и (C60Pd2)n. Подобные структуры получили название „нить жемчуга“.
Фуллерит пока не находит широкого практического применения ввиду своей малоизученности и большой стоимости, но уже сейчас ведутся интенсивные работы по применению этого вещества в различных областях медицины и техники. Недавно было показано, что фуллерит при давлении всего лишь 2·105 атм и комнатной температуре переходит в алмаз, что чрезвычайно упрощает получение этого ценного материала. Обсуждается также возможное применение эндоэдральных соединений фуллеренов с радиоактивными изотопами металлов для лечения раковых заболеваний. В 1994 году фирма „Мицубиси“ начала выпуск аккумуляторов на базе фуллерита, действие которых, подобно обычным металлогидридным, основано на присоединении водорода. Фуллеритовый аккумулятор имеет в 5 раз большую емкость, чем обычный, и, кроме того, более легкий и экологически безопасный. Их планируют использовать для питания персональных компьютеров и слуховых аппаратов. Обсуждаются возможности применения фуллеренов для создания алмазоподобных пленок, фотоприемников, лекарственных препаратов, сверхпроводников, а также их применение в качестве красителей для копировальных машин.
ДИОКСИД УГЛЕРОДА.
В лабораторных условиях диоксид углерода удобно получать действием соляной кислоты на известняк или мрамор. Молекула О=С=О линейна. Диоксид углерода — бесцветный газ со слегка кисловатым запахом и вкусом. Под давлением около 60 атм он сжижается уже при обычных температурах в бесцветную жидкость (её хранят и перевозят в стальных баллонах). При сильном охлаждении он застывает в белую снегообразную массу, под обычным давлением возгоняется при -78 °С. Предварительно спрессованный твёрдый СО2 испаряется довольно медленно, причём окружающее пространство сильно охлаждается. На этом основано его применение в качестве “сухого льда”.
Углекислый газ был впервые описан Ван Гельмонтом. Большие его количества получаются как побочный продукт при обжиге известняка и некоторых других процессах (сжигание кокса, спиртовое брожение и т. д.). Иногда в качестве источника диоксида углерода используют обычные топочные газы, пропуская их (после освобождения от твёрдых частиц дыма и охлаждения) сквозь концентрированный раствор поташа, хорошо поглощающий диоксид углерода на холоду и вновь выделяющий его при нагревании.
Ядерное расстояние d(С-О) = 116 пм; оно существенно меньше, чем обычно для двойной связи d(C=O) = 123 пм. Этому соответствует и высокое значение средней энергии связи (803 кДж/моль). По-видимому, в молекуле диоксида имеется некоторое повышение порядка валентных связей за счёт свободных электронов атомов кислорода.
Баллоны для хранения жидкого СО2 должны иметь чёрную окраску и жёлтую надпись “Углекислота”. При давлении 35 тыс. атм твёрдый СО2 становится хорошим проводником тока (причём по мере повышения температуры электропроводность его возрастает).
Твёрдый СО2 используется также при проведении взрывных работ на угольных разработках. Помещённый поверх взрывного вещества “сухой лёд” мгновенно испаряется от теплоты взрыва с образованием большого объёма диоксида. Тем самым, с одной стороны, расширяется полезная площадь взрыва и улучшается качество выброшенного угля (т.к. он меньше крошится), с другой — предотвращается возможность вторичных взрывов при наличии в пласте воспламеняющихся газов.
Твёрдый диоксид углерода можно использовать для устранения облачности над аэродромами. Облака состоят из мельчайших капелек переохлаждённой воды. Нарушение их метастабильного состояния с выделением дождя или снега (в зависимости от погоды) хорошо достигается рассеиванием над облаками измельчённого до определённых размеров твёрдого диоксида углерода. Каждая его крупинка, имеющая температуру около -80 °С, при падении сквозь облако вызывает кристаллизацию соседних капелек, создавая тем самым громадное число зародышевых снежинок. Так как давление водяного пара над ними ниже, чем над переохлаждённой водой, эти снежинки растут за счёт капелек и затем оседают вниз. Устранение облачности осуществляется примерно за полчаса, причём для осаждения одного кубического километра облака (содержащего до 1000 т воды) требуется лишь около 200 г сухого льда. Этот метод можно использовать и для искусственного дождевания посевов.
Примерно на 97% из СО2 состоит атмосфера Венеры. Существование атмосферы у этой планеты было открыто М. В. Ломоносовым (1761 г.). Наблюдая её прохождение по диску Солнца, он установил, что Венера “окружена знатною воздушною атмосферой, таковою (лишь бы не большей), какова обливается около нашего шара земного”. Атмосферное давление на поверхности Венеры (нагретой до 475 °С) составляет около 90 атм. Весьма разрежённая атмосфера Марса также состоит главным образом из диоксида углерода.
Углекислый газ (иначе “углекислота”) не поддерживает горения обычных видов топлива (т.е. углерода и его соединений). Горят в углекислом газе лишь такие вещества, сродство которых к кислороду значительно больше, чем у углерода. Примером может служить металлический магний, около 600 °С загорающийся в углекислом газе и сгорающий по уравнению:
СО2 + 2 Mg = 2 MgO + C + 811 кДж.
Атмосфера содержит в среднем 0,03% СО2 по объёму.
Углекислый газ не поддерживает жизнедеятельности бактерий и плесеней, поэтому сроки сохраняемости пищевых продуктов в атмосфере этого газа увеличиваются. С другой стороны, повышенное содержание СО2 в воздухе теплиц ведёт к стимулированию роста растений (“углекислотное удобрение”). Практически это достигается путём помещения в теплицы кусков сухого льда. Для большинства овощных культур наиболее благоприятным оказалось содержание СО2 от 0,2 до 0,3%.
На организм человека концентрация углекислого газа в воздухе до 3% вредного влияния не оказывает. Наблюдается лишь учащённое дыхание в результате стимулирующего воздействия растворённого в крови СО2 на соответствующие центры нервной системы. Вдыхание СО2 в более высоких концентрациях ведёт к серьёзным расстройствам работы организма. При 10%-ной концентрации быстро наступает потеря сознания и смерть вследствие остановки дыхания, а 20%-ная концентрация вызывает паралич жизненных центров в течение нескольких секунд. Смесь кислорода с 5% СО2 (“карбоген”) находит медицинское использование при задержке дыхания и некоторых отравлениях.
В воде СО2 растворим довольно хорошо (приблизительно 1:1 по объёму).
УГОЛЬНАЯ КИСЛОТА И ЕЁ СОЛИ.
При растворении углекислого газа происходит его частичное взаимодействие с водой, ведущее к образованию угольной кислоты:
Н2О + СО2Ы Н2СО3.
Хотя равновесие этой реакции сильно смещено влево, СО2 следует считать ангидридом угольной кислоты. Последняя очень слаба и лишь незначительно распадается на ионы Н· и НСО3’, а дальнейшая её диссоциация с образованием ионов СО3” сама по себе почти не идёт. Учитывая, однако, возможность и такой диссоциации, можно написать следующее равновесие в водном растворе СО2:
Н2О + СО2Ы Н2СО3 Ы Н·+ НСО3’ Ы 2 Н· + СО3”.
При нагревании СО2 улетучивается и равновесие смещается влево; напротив, при прибавлении щёлочи происходит связывание ионов водорода и смещение равновесия вправо.
Растворимость СО2 в воде составляет (по объёму): 1,71 при 0 °С; 0,88 при 20 °С; 0,36 при 60 °С. В равновесии с воздухом в воде содержится около 5·10-4 г/л диоксида и за счёт его растворения она приобретает рН = 5,7, а насыщенный при обычных условиях водный раствор является приблизительно 0,04 М относительно СО2 и имеет рН = 3,7. Из насыщенного на холоду раствора может быть выделен кристаллогидрат СО2·6Н2О, являющийся аддуктом. Вместе с тем для угольной кислоты известен устойчивый лишь ниже 5 °С эфират (С2Н5)2О·Н2СО3 (т. пл. -47 °С).
С хорошей растворимостью углекислого газа связано его использование при изготовлении искусственных минеральных вод. Из них обычная газированная вода представляет собой просто насыщенный водный раствор СО2, а в состав других входят, кроме того, примеси некоторых солей. Подобным же образом готовят и “прохладительные напитки” (лимонад и др.) с той лишь разницей, что вместо солей добавляют небольшие количества сахара и различных “эссенций”.
При оценке силы Н2СО3 (К1 = 4·10-7, К2 = 5·10-11), имеющуюся в растворе концентрацию Н• относят к общему количеству растворённого СО2 (допуская тем самым, что он весь находится в виде Н2СО3). Между тем, по сути дела, следовало бы исходить из концентрации действительно имеющихся в растворе молекул Н2СО3. Так как последних мало, угольная кислота должна быть значительно более сильной, чем это нам представляется. Попытки оценить её истинную константу диссоциации приводят к значению К1 = 2·10-4, т.е. в 500 раз превышающему непосредственно определяемое. Подобные же соображения применимы и к другим кислотам и основаниям (сернистой кислоте, гидроксиду аммония и др.), заметно распадающимся в растворе не только на ионы, но и на нейтральные молекулы. Во всех этих случаях истинные константы диссоциации должны быть выше непосредственно определяемых.
Молекулы Н2СО3 могут образовываться и в газовой фазе. На это указывает несравненно более сильное повышение растворимости водяного пара по мере роста давления углекислого газа сравнительно, например, с азотом.
Будучи двухосновной кислотой, Н2СО3 даёт два ряда солей: средние (с анионом СО3”) и кислые (с анионом НСО3’). Первые называются карбонатами, вторые гидрокарбонатами или бикарбонатами. Подобно самим анионам угольной кислоты, большинство её солей бесцветно.
Из карбонатов наиболее обычных катионов растворимы только соли Na, K и NH4. В результате значительного гидролиза их растворы имеют щелочную реакцию. Карбонаты натрия и калия плавятся без разложения, а большинство остальных карбонатов при прокаливании разлагаются на углекислый газ и оксид соответствующего металла. Под действием сильных кислот все карбонаты легко разлагаются с образованием соли сильной кислоты, воды и углекислого газа. Наиболее практически важны карбонат натрия (сода), карбонат калия (поташ) и карбонат кальция (мел, мрамор, известняк).
В противоположность большинству карбонатов, все бикарбонаты в воде растворимы. Наиболее важной кислой солью угольной кислоты является гидрокарбонат натрия (“двууглекислая” или “питьевая” сода). Гидролиз её при обычных условиях незначителен (реакция раствора на лакмус почти нейтральна). При нагревании он заметно увеличивается, а около 60 °С углекислый газ начинает частично выделяться из раствора.
Содержащийся в карбонатах ион СО32- имеет структуру равностороннего треугольника с атомом С в центре (d(CO) = 129 пм). В ионе НСО3- для двух атомов кислорода d(CO) = 126 пм (с углом 125° между ними), тогда как для третьего d(СО) = 135 пм.
Одно из важных применений NaHCO3 связано с изготовлением огнетушителей. Пламя может быть потушено одним из следующих способов (или их комбинированием): 1) удалением горючего материала, 2) прекращением доступа кислорода и 3) охлаждением горящего вещества ниже его температуры воспламенения. Огнетушители с NaHCO3 работают по второму и отчасти третьему способам. Баллон заполнен крепким раствором NaHCO3 (с примесью веществ, способствующих образованию пены). В верхней части баллона имеется стеклянная ампула, содержащая серную кислоту. Для приведения огнетушителя в действие его переворачивают вверх дном и разбивают ампулу помещённым в крышке ударником. Кислота вступает в соприкосновение с раствором NaHCO3, причём тотчас образуется большое количество углекислого газа. Насыщенная им жидкость вытекает сильной струёй и покрывает горящее место густой пеной. Последняя охлаждает его (за счёт испарения воды), главным же образом изолирует от кислорода воздуха, благодаря чему прекращается горение.
Иногда для тушения огня пользуются небольшими баллонами с жидким СО2. При испарении последнего горящее вещество одновременно охлаждается (за счёт испарения СO2) и изолируется от кислорода воздуха слоем углекислого газа. Главное преимущество огнетушителей этого типа заключается в том, что СО2 испаряется без остатка, и предметы окружающие место горения не портятся.
Перексидные соединения углерода производятся от неизвестных в свободном состоянии надугольной и мононадугольной кислот: НООС-О-О-СООН и НООС-О-О-Н. Соли надугольной кислоты (надуглекислые, или перкарбонаты) известны для Na, K и Rb. Они образуются в результате анодного окисления концентрированных растворов карбонатов при низких температурах (по схеме: 2СО3” - 2е-= С2О6”) и представляют собой бесцветные (или бледно-синеватые) кристаллические вещества, чрезвычайно гигроскопичные, но в сухом состоянии устойчивые. При нагревании эти соли переходят в карбонаты с выделением углекислого газа и кислорода, при растворении подвергаются гидролизу:
К2С2О6 + 2 Н2О Ы 2 КНСО3 + Н2О2 .
При действии на них кислот выделяющаяся Н2С2О6 тотчас распадается на Н2О2 и СО2.
Для мононадугольной кислоты известны не только средние (как для надугольной), но и кислые соли. Те и другие могут быть получены взаимодействием с углекислым газом пероксидов или гидропероксидов щелочных металлов по схемам:
Э2О2 + СО2 = Э2СО4 или ЭООН + СО2 = ЭНСО4.
По свойствам они похожи на соли надугольной кислоты. Известны также продукты присоединения пероксида водорода к карбонатам, например “персоль” Na2CO3·1,5Н2О2·Н2О. Являются ли пероксидные производные угольной кислоты продуктами присоединения или истинными солями надугольных кислот, до конца не ясно. Надуглекислый калий (К2С2О6) применяется иногда в качестве окислителя при химических анализах. Из нейтрального раствора КI он тотчас выделяет свободный иод.
Кроме воды, СО2способен присоединять аммиак уже при обычных условиях с образованием карбаминовокислого аммония:
СО2+ 2 NH3= CO(NH2)ONH4.
Эта нестойкая соль неизвестной в свободном состоянии аминоугольной кислоты при нагревании до 190 °С под давлением около 200 атм отщепляет молекулу воды и переходит в карбамид:
СО(NH2)ONH4+ 29 кДж = Н2О + СО(NH2)2.
Карбамид (иначе мочевина) представляет собой бесцветные кристаллы (т. пл. 133 °С), хорошо растворимые в воде (приблизительно 1:1 по массе при обычных условиях). В расплавленном состоянии он является хорошим растворителем многих неорганических веществ, а с солями ряда металлов способен образовывать комплексные соединения. Взаимодействие карбамида с пиросерной кислотой по реакции:
СО(NH2)2+ H2S2O7= CO2+ 2 H2NSO3H
является удобным методом получения сульфаминовой кислоты.
Основные свойства карбамида выражены крайне слабо (К = 2·10-14), но соли его с кислотами получены. Примерами могут служить СО(NH2)2·HNO3и СО(NH2)2·H3PO4.
Карбамид постоянно содержится в моче животных (отсюда его другое название). Он является прекрасным азотным удобрением и хорошим частичным заменителем растительных белковых кормов для жвачных животных (но ядовит для остальных).
В водном растворе (и особенно в почве под воздействием бактерий) карбамид медленно присоединяет две молекулы воды и переходит в углекислый аммоний:
СО(NH2)2+ 2 H2O = (NH4)2CO3.
Именно этой реакцией обусловлен аммиачный (вследствие последующего гидролиза (NH4)2CO3) запах в плохо содержимых отхожих местах, зверинцах и т. п. С мочой взрослого человека в сутки выделяется около 25 г мочевины.
УГАРНЫЙ ГАЗ.
Монооксид углерода образуется при сгорании углерода в недостатке кислорода. Чаще всего он получается в результате взаимодействия углекислого газа с раскалённым углём:
СО2+ С + 171 кДж = 2 СО.
Реакция эта обратима, причём равновесие её ниже 400 °С практически нацело смещено влево, а выше 1000 °С — вправо (рис. 7). Однако с заметной скоростью оно устанавливается лишь при высоких температурах. Поэтому в обычных условиях СО вполне устойчив.
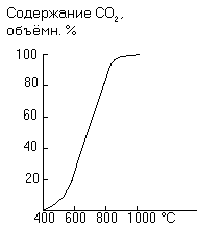
Рис. 7. Равновесие СО2+ С Ы2 СО.
Молекула СО характеризуется d(СО) = 113 пм, энергия его диссоциации 1070 кДж/моль, что больше, чем у других двухатомных молекул. Рассмотрим электронное строение СО, где атомы связаны между собой двойной ковалентной связью и одной донорно-акцепторной, причём кислород является донором, а углерод акцептором.
Монооксид углерода (т. пл. -205 °С, т. кип. -191 °С) входит в состав атмосферы (10-5объёмн. %). В среднем 0,5% СО содержит табачный дым и 3% — выхлопные газы двигателей внутреннего сгорания. Образование СО из элементов идёт по уравнению:
2 С + О2= 2 СО + 222 кДж.
Критическая температура СО -140°С, критическое давление 35 атм. Растворимость СО в воде около 1:40 по объёму.
Небольшие количества СО удобно получать разложением муравьиной кислоты:
НСООН = Н2О + СО
Реакция эта легко протекает при взаимодействии НСООН с горячей крепкой серной кислотой. Практически это получение осуществляют либо действием конц. серной кислоты на жидкую НСООН (при нагревании), либо пропусканием паров последней над гемипентаоксидом фосфора. Взаимодействие НСООН с хлорсульфоновой кислотой по схеме:
НСООН + СISO3H = H2SO4+ HCI + CO
идёт уже при обычных температурах.
Удобным методом лабораторного получения СО могут служить нагревание с конц. серной кислотой щавелевой кислоты или железосинеродистого калия. В первом случае реакция протекает по схеме:
Н2С2О4 = СО + СО2+ Н2О.
Наряду с СО выделяется и углекислый газ, который может быть задержан пропусканием газовой смеси сквозь раствор гидроксида бария. Во втором случае единственным газообразным продуктом является оксид углерода:
К4[Fe(CN)6] + 6 H2SO4+ 6 H2O = 2 K2SO4+ FeSO4+ 3 (NH4)2SO4+ 6 CO.
Монооксид углерода представляет собой бесцветный и не имеющий запаха газ, малорастворимый в воде и химически с ней не взаимодействующий. Не реагирует СО также со щелочами и кислотами. Он чрезвычайно ядовит.
Первыми признаками острого отравления СО являются головная боль и головокружение, в дальнейшем наступает потеря сознания. Предельно допустимая концентрация СО в воздухе промышленных предприятий считается 0,02 мг/л. Основным противоядием при отравлении СО служит свежий воздух. Полезно также кратковременное вдыхание паров нашатырного спирта.
Опытами на молодых крысах было установлено, что содержание в воздухе 0,02% СО замедляет их рост и снижает активность по сравнению с контрольными экземплярами. Особенно интересным оказалось следующее наблюдение: животным обеих партий предоставлялись на выбор вода, раствор глюкозы и раствор спирта. Крысы, живущие в обычном воздухе, предпочитали воду, а живущие в атмосфере СО — раствор спирта.
Чрезвычайная ядовитость СО, отсутствие у него цвета и запаха, а также очень слабое поглощение его активированным углём обычного противогаза делают этот газ особенно опасным. Вопрос защиты от него был разрешён изготовлением специальных противогазов, коробка которых заполнялась смесью различных оксидов (в основном MnO2и CuO). Действие этой смеси (“гопкалита”) сводится к каталитическому ускорению реакции окисления СО до СО2кислородом воздуха. На практике гопкалитовые противогазы очень неудобны, так как заставляют дышать нагретым (в результате реакции окисления) воздухом.
С химической стороны монооксид углерода характеризуется главным образом склонностью к реакциям присоединения и своими восстановительными свойствами. Однако обе эти тенденции обычно проявляются лишь при повышенных температурах. В этих условиях СО соединяется с кислородом, хлором, серой, некоторыми металлами и т. д. Вместе с тем оксид углерода при нагревании восстанавливает до металлов многие оксиды, что весьма важно для металлургии.
Наряду с нагреванием повышение химической активности СО часто вызывается его растворением. Так, в растворе он способен восстанавливать соли Au, Pt и некоторых других элементов до свободных металлов уже при обычных температурах.
При повышенных температурах и высоких давлениях имеет место взаимодействие СО с водой и едкими щелочами: в первом случае образуется НСООН, а во втором — муравьинокислый натрий. Последняя реакция протекает при 120 °С, давлении 5 атм и находит техническое использование.
Легко идущее в растворе восстановление хлористого палладия по суммарной схеме:
PdCl2+ H2O + CO = CO2+ 2 HCl + Pd
служит наиболее часто применяемой реакцией открытия монооксида углерода в смеси газов. Уже очень небольшие количества СО легко обнаруживаются по лёгкому окрашиванию раствора вследствие выделения мелко раздробленного металлического палладия. Количественное определение СО основывается на реакции:
5 СО + I2O5 = 5 CO2 + I2.
Окисление СО в растворе часто идёт с заметной скоростью лишь в присутствии катализатора. При подборе последнего основную роль играет природа окислителя. Так, KMnO4 быстрее всего окисляет СО в присутствии мелкораздробленного серебра, K2Cr2O7 — в присутствии солей ртути, КСlO3 — в присутствии OsO4. В общем, по своим восстановительным свойствам СО похож на молекулярный водород, причём активность его при обычных условиях выше, чем у последнего. Интересно, что существуют бактерии, способные за счёт окисления СО получать необходимую им для жизни энергию.
Сравнительную активность СО и Н2 как восстановителей можно оценить путём изучения обратимой реакции:
Н2О + СО Ы СО2 + Н2 + 42 кДж,
равновесное состояние которой при высоких температурах устанавливается довольно быстро (особенно в присутствии Fe2O3). При 830 °С в равновесной смеси находятся равные количества СО и Н2, т. е. сродство обоих газов к кислороду одинаково. Ниже 830 °С более сильным восстановителем является СО, выше — Н2.
Связывание одного из продуктов рассмотренной выше реакции в соответствии с законом действия масс смещает её равновесие. Поэтому, пропуская смесь монооксида углерода и водяного пара над оксидом кальция, можно получить водород по схеме:
Н2О + СО + СаО = СаСО3 + Н2 + 217 кДж.
Реакция эта идёт уже при 500 °С.
На воздухе СО загорается около 700 °С и сгорает синим пламенем до СО2:
2 СО + О2 = 2 СО2 + 564 кДж.
Сопровождающее эту реакцию значительное выделение тепла делает монооксид углерода ценным газообразным топливом. Однако наиболее широкое применение он находит как исходный продукт для синтеза различных органических веществ.
Сгорание толстых слоёв угля в печах идёт в три стадии:
1) С + О2 = СО2 ; 2) СО2 + С = 2 СО; 3) 2 СО + О2 = 2 СО2.
При преждевременном закрытии трубы в печи создаётся недостаток кислорода, что может вызвать распространение СО по отапливаемому помещению и привести к отравлениям (угар). Следует отметить, что запах “угарного газа” обусловлен не СО, а примесями некоторых органических веществ.
Пламя СО может иметь температуру до 2100 °С. Реакция горения СО интересна тем, что при нагревании до 700-1000 °С она идёт с заметной скоростью только в присутствии следов водяного пара или других содержащих водород газов (NH3, H2S и т. п.). Обусловлено это цепным характером рассматриваемой реакции, протекающей при посредстве промежуточного образования радикалов ОН по схемам:
Н + О2 = НО + О, затем О + СО = СО2, НО + СО = СО2 + Н и т. д.
При очень высоких температурах реакция горения СО становится заметно обратимой. Содержание СО2 в равновесной смеси (под давлением 1 атм) выше 4000 °С может быть лишь ничтожно малым. Сама молекула СО настолько термически устойчива, что не разлагается даже при 6000 °С. Молекулы СО были обнаружены в межзвёздной среде.
Большие количества СО могут быть получены путём неполного сжигания каменного угля в специальных печах — газогенераторах. Обычный (“воздушный”) генераторный газ содержит в среднем (объёмн. %): СО-25, N2-70, СО2-4 и небольшие примеси других газов. При сжигании он даёт 3300-4200 кДж на м3. Замена обычного воздуха на кислород ведёт к значительному повышению содержания СО (и увеличению теплотворной способности газа).
Ещё больше СО содержит водяной газ, состоящий (в идеальной случае) из смеси равных объёмов СО и Н2 и дающий при сгорании 11700 кДж/м3. Газ этот получают продувкой водяного пара сквозь слой раскалённого угля, причём около 1000 °С имеет место взаимодействие по уравнению:
Н2О + С + 130 кДж = СО + Н2.
Реакция образования водяного газа идёт с поглощением тепла, уголь постепенно охлаждается и для поддержания его в раскалённом состоянии приходится пропускание водяного пара чередовать с пропусканием в газогенератор воздуха (или кислорода). В связи с этим водяной газ содержит приблизительно СО-44, Н2-45, СО2-5 и N2-6%. Он широко используется для синтезов различных органических соединений.
Часто получают смешанный газ. Процесс его получения сводится к одновременному продуванию сквозь слой раскалённого угля воздуха и паров воды, т.е. комбинированию обоих описанных выше методов — Поэтому состав смешанного газа является промежуточным между генераторным и водяным. В среднем он содержит: СО-30, Н2-15, СО2-5 и N2-50%. Кубический метр его даёт при сжигании около 5400 кДж.
Перечисленные выше газы используются в качестве топлива и исходного сырья химической промышленности. Они важны, например, как один из источников получения азотно-водородной смеси для синтеза аммиака. При пропускании их совместно с водяным паром над нагретым до 500 °С катализатором (главным образом Fe2O3) происходит взаимодействие по обратимой реакции:
Н2О + СО Ы СО2 + Н2 + 42 кДж,
равновесие которой сильно смещено вправо. Образовавшийся углекислый газ удаляют затем промыванием водой (под давлением), а остаток СО — аммиачным раствором солей меди. В результате остаются почти чистый азот и водород. Соответственно регулируя относительные количества генераторного и водяного газов, можно получать N2 и Н2 в требуемом объёмном соотношении. Перед подачей в колонну синтеза газовую смесь подвергают сушке и очистке от отравляющих катализатор примесей.
При действии СО на металлический К при 80 °С образуется бесцветное кристаллическое очень взрывчатое соединение состава К6С6О6. Вещество это с отщеплением калия легко переходит в оксид углерода С6О6 (“трихинон”), который можно рассматривать как продукт полимеризации СО. Строение его отвечает шестичленному циклу, образованному атомами углерода, каждый из которых соединён двойной связью с атомами кислорода.
Ещё один оксид углерода (“недоокись”) состава С3О2 может быть получен отнятием воды от малоновой кислоты СН2(СООН)2 при помощи Р2О5. Теплота его образования из элементов 96 кДж/моль. Это бесцветный газ с резким запахом (т. пл. -107 °С, т. кип. +7 °С). Строениеего молекулы отвечает линейной структуре О=С=С=С=О с d(CO)=116, d(CC)=129 пм. При нагревании С3О2 легко полимеризуется с образованием красного полимера и почти так желегко разлагается на СО2 и С2 (с дальнейшим переходом молекул углерода в графит). На воздухе он горит синим пламенем с выделением копоти, а при взаимодействии с водой даёт малоновую кислоту.
СОS. Взаимодействие СО с серой по реакции:
СО + S = COS + 29 кДж
быстро идёт лишь при высоких температурах. Образующийся тиооксид углерода (О=С=S) представляет собой бесцветный и не имеющий запаха газ (т. пл. -139, т. кип. -50 °С). В воде он довольно хорошо растворим (1:2 по объёму) и постепенно гидролизуется по схеме:
СОS + H2O = CO2 + H2S.
Молекула ОСS линейна и полярна. Известны аналогичные производные Se и Те.
COCl2. Взаимодействие СО с хлором по уравнению:
СО + Сl2 Ы СОСl2 + 113 кДж
в присутствии катализатора (активированного угля) довольно быстро идёт уже при комнатной температуре. Получающийся фосген представляет собой бесцветный, очень ядовитый газ с характерным запахом, малорастворимый в воде, но постепенно разлагающийся ею по схеме:
СОСl2 + 2 Н2О = Н2СО3 + 2 НСl
Он является, следовательно, хлорангидридом угольной кислоты. Ввиду большой реакционной способности фосген находит широкое использование при органических синтезах.
Молекула ОССl2 полярна, имеет плоское строение и характеризуется следующими структурными параметрами: d(СО) = 117, d(ССI) = 175 пм, РСlССl = 111°. Как растворитель фосген (т. пл. -128, т. кип. +8 °С) малоактивен — растворяет лишь немногие неорганические вещества ковалентного характера (I2, ICl, AlCl3, AsCl3, SbCl3, SbCl5, хлориды серы). Растворы в нём хлористого алюминия хорошо проводят электрический ток и обладают большой реакционной способностью. Причиной этого является наличие равновесия по схеме:
СОСl2 + AlCl3 Ы СОСl++ АlCl4-.
Чрезвычайная ядовитость фосгена наряду с его большой плотностью по отношению к воздуху, дешевизна и лёгкость получения обусловили применение этого газа в первую мировую войну как боевого отравляющего вещества. При отравлении им необходимо предоставить пострадавшему полный покой. Полезно также вдыхание чистого кислорода. Предельно допустимой концентрацией фосгена в воздухе промышленных предприятий считается 5·10 -4 мг/л. Содержащие его баллоны должны иметь окраску защитного цвета с красной полосой.
Аналогичный фосгену фторид — СОF2 (т. пл. -114, т. кип. -83 °С) — образуется из СО и F2 c большим выделением тепла (481 кДж/моль). Молекула его полярна и характеризуется параметрами d(СО) = 117, d(CF) = 131 пм, РFCF = 108°.
Монооксид углерода (II) способен непосредственно соединяться с некоторыми металлами. В результате образуются карбонилы металлов [Fe(CO)5, Ni(CO)4, Mo(CO)6 и др.], которые следует рассматривать как комплексные соединения.
Карбонилы металлов представляют собой летучие жидкие или твёрдые вещества, нерастворимые в воде, но хорошо растворяющиеся во многих органических растворителях. Все они весьма ядовиты, а при нагревании легко распадаются на соответствующий металл и оксид углерода(II).
Карбонильные производные известны для многих металлов, являющихся d-элементами середин больших периодов. Получают их обычно под высоким давлением СО и при нагревании, исходя либо из мелко раздробленных металлов, либо из их соединений, восстанавливающихся до металлов в процессе самого синтеза.
Наиболее давно известны карбонилы хрома и его аналогов. Они отвечают формуле Э(СО)6 и представляют собой бесцветные, легко возгоняющиеся кристаллы (т. возгонки соответственно 147, 156 и 175 °С). Под уменьшенным давлением они могут быть возогнаны без разложения, а под обычным давлением около 120 °С начинают медленно разлагаться на металл и СО.
Хотя карбонилы Мо и W могут быть получены прямым синтезом, чаще исходят из МоСl5 и WCl6, а Сr(CO)6 обычно получают исходя из CrCl3. Молекулы всех трёх соединений представляют собой правильные октаэдры.
По химической стойкости рассматриваемые карбонилы превосходят все другие соединения этого типа. При обычной температуре на них не действуют ни концентрированные НСl и H2SO4, ни щёлочи (в отсутствие кислорода). Однако дымящей азотной кислотой они легко разрушаются. Под действием хлора происходит полное отщепление СО с образованием хлоридов соответствующих металлов.
Известны разнообразные продукты частичного замещения СО в карбонилах Э(СО)6 (главным образом на различные амины, причём обычно замещается не более трёх молекул СО). Например, взаимодействие суспензии Cr(CO)6 с металлическим натрием в жидком аммиаке идёт по реакции:
Сr(CO)6 + 2 Na = Na2[Cr(CO)5] + CO.
Образующийся продукт представляет собой жёлтое твёрдое вещество, устойчивое в атмосфере азота, а на воздухе быстро окисляющееся. Аналогичные производные получены для Мо и W. Описаны также жёлтые соединения рассматриваемых элементов типа Na2[Э2(СО)10] и чёрные типа Na2[Э3(СО)14] (где Э — Сr или Мо). Раствор Na2[Cr(CO)5] в жидком аммиаке устойчив, но под действием солей аммония протекает реакция:
Na2[Cr(CO)5] + 2 NH4X = 2 NaX + H2 + NH3 + Cr(CO)5NH3.
Взаимодействие карбонилов Э(СО)6 всех трёх элементов с NH3 при нагревании ведёт к образованию Э(СО)5NH3. Твёрдый жёлтый Сr(CO)5NH3 хорошо растворим в ряде органических растворителей и медленно разлагается на воздухе.
Карбонилы Мn, Тс и Re могут быть получены исходя из солей двухвалентного марганца или высших оксидов Тс и Re. Это летучие кристаллические вещества, образованные молекулами Э2(СО)10. В структуре последних каждый центральный атом Э соединён с пятью молекулами СО ( d(ReC) = 201 пм) и с другим атомом Э длинной связью Э–Э (293 пм у Мn и 304 пм у Тс и Re). Таким образом, атомы Э оказываются приблизительно в центрах октаэдров, примерно на 45° повёрнутых относительно друг друга. Получен и жёлтый смешанный карбонил (СО)5МnRe(CO)5 со связью Мn-Re (296 пм). В отличии от своих бесцветных аналогов Мn2(СО)10 имеет золотисто-жёлтую окраску.
Известны многочисленные производные карбонилов марганца и рения. При разрыве связей Э-Э в карбонилах Э2(СО)10 получаются соединения типа ХЭ(СО)5, где Х – одновалентный атом или радикал, занимающий один из углов октаэдра около атома Э. Так, взаимодействием Э2(СО)10 с амальгамой натрия могут быть получены желтоватые, энергично окисляющиеся на воздухе соли NaЭ(CO)5, а из них легко идущим гидролизом — гидрокарбонилы НЭ(СО)5.
Последние представляют собой бесцветные жидкости с т. пл. -25 (Мn) или +13 °С (Re), малорастворимые в воде, но смешивающиеся со многими органическими растворителями. Разложение их на Э2(СО)10 и Н2 идёт при обычных условиях крайне медленно.
При действии на карбонилы Э2(СО)10 галогенов образуются карбонилгалогениды ГЭ(СО)5 (где Г — Cl, Br, I). Они представляют собой довольно устойчивые бесцветные или желтоватые кристаллические вещества нерастворимые в воде. Их летучесть и растворимость в органических жидкостях возрастает по ряду Сl–Br–I. При нагревании галогениды ГЭ(СО)5 отщепляется часть СО и переходят в димерные галогенкарбонилы [ГЭ(СО)4]2, структура которых отвечает двум октаэдрам с общим ребром из атомов галогена. В ряду Cl–Br–I такой переход облегчается. Образующиеся бесцветные или жёлтые вещества плохо растворимы в органических жидкостях.
Подобно карбонилам Cr, Mo и W, твёрдый при обычных условиях зелёный V(CO)6 имеет структуру правильного октаэдра с атомом ванадия в центре. Вещество это на воздухе самовоспламеняется. Очень сильное охлаждение вызывает его димеризацию с образованием V2(CO)12. Растворы V(CO)6 в органических растворителях имеют жёлто-оранжевый цвет и очень неустойчивы. При действии на них иода количественно протекает реакция по схеме:
2 V(CO)6 + 3 I2 = 2 VI3 + 12 CO.
С другой стороны, V(CO)6 легко восстанавливается до аниона [V(CO)6]–, для которого известны, в частности, жёлтые соли типа М[V(CO)6], где М = Na, K, NH4.
Оксид углерода(II) образует комплексные соединения также с некоторыми солями. Одни из них (OsCl2·3CO, PtCl2·CO и т. д.) устойчивы только в растворе. С образованием последнего вещества связано поглощение оксида углерода(II) раствором СuСl в крепкой НСl. Подобные же соединения образуются, по-видимому, и в аммиачном растворе CuCl, часто применяемом для поглощения СО при анализе газов.
СЕРОУГЛЕРОД.
В технике сероуглерод (СS2) получают пропусканием паров серы сквозь слой раскалённого угля.
В противоположность сильно экзотермическому процессу образования СО2 из элементов, реакция соединения углерода с серой является эндотермической:
С + 2 S + 88 кДж = СS2.
Чистый сероуглерод представляет собой весьма летучую бесцветную жидкость с довольно приятным запахом, но обычно он содержит незначительные примеси продуктов частичного разложения, сообщающие ему жёлтый цвет и отвратительный запах. В воде сероуглерод почти нерастворим и при обычных условиях с ней не взаимодействует. Пары его ядовиты и очень легко воспламеняются. Сгорание их идёт по уравнению:
СS2 + 3 O2 = CO2 + 2 SO2 + 1100 кДж.
Сероуглерод является прекрасным растворителем жиров, масел, смол и т. п. На этом основано его применение для экстрагирования (извлечения) подобных веществ из различных природных материалов. Он имеет большое значение для промышленности искусственных волокон и используется также для борьбы с вредителями сельского хозяйства.
Молекула S=C=S линейна, d(C=S) = 156 пм и энергия 535 кДж/моль.
Растворимость сероуглерода (т. пл. -112, т. кип. 46 °С) в воде составляет всего 0,15 вес. %. Гидролиз его по схеме:
СS2 + 2 H2O = CO2 + 2 H2S + 50 кДж.
протекает лишь выше 150 °С. Ежегодная мировая выработка сероуглерода составляет около 1 млн. т.
Вдыхание воздуха с содержанием 0,3% СS2 и выше может быстро привести к тяжёлому заболеванию. При хроническом отравлении малыми дозами паров сероуглерода постепенно развиваются желудочные заболевания (ахилия, гастрит) и различные расстройства нервной системы. Предельно допустимой концентрацией СS2 в воздухе промышленных предприятий считается 0,01 мг/л. Смеси паров сероуглерода с воздухом взрывчаты при содержании от 1 до 50 объёмн. % СS2.
Под давлением 45 тыс. атм сероуглерод при 200 °С превращается в чёрную твёрдую полимерную массу с плотностью 1,9 г/см3. Полимеризация идёт, с образованием цепей типа [-C(S)-S-]n и сопровождается выделением тепла (23,4 кДж/моль СS2). Полимер имеет низкую диэлектрическую проницаемость (4,0), обладает полупроводниковыми свойствами и нерастворим в органических растворителях. В обычных условиях он устойчив, но при 70 °С размягчается, а при 170 °С разлагается на элементы.
Наряду с сероуглеродом известны сернистые аналоги и других кислородных соединений углерода – монотиооксид (СS) и тионедооксид (С3S2). Первый образуется в виде чрезвычайно неустойчивого твёрдого бесцветного вещества под действием тлеющего электрического разряда на пары СS2 при очень низких температурах. Второй несколько более устойчив, может быть получен нагреванием паров СS2 в пламени электрической дуги. Недооксид углерода (S=C=C=C=S) представляет собой красную жидкость с острым запахом, затвердевающую при -1 °С. Как СS так и C3S2 очень легко самопроизвольно превращаются в темноокрашенные твёрдые продукты полимеризации. Аналогичные производные дают Se и Те.
Подобно СО2,сероуглерод является кислотным ангидридом и с некоторыми сульфидами может образовывать соли тиоугольной кислоты (Н2СS3). Так, при взаимодействии СS2 с крепким раствором Na2S образуется Na2CS3. Тиокарбонаты наиболее активных металлов (а также NH4+) устойчивы, тогда как производные остальных более или менее легко разлагаются. В твёрдом виде большинство солей Н2СS3 жёлтого цвета, а растворы их обычно имеют красную окраску. Хорошо растворимы в воде лишь немногие тиокарбонаты, в частности, производные Na, K и NH4. Тиокарбонат калия применяется для борьбы с вредителями сельского хозяйства (главным образом с филлоксерой).
Свободная тиоугольная кислота может быть получена действием сильных кислот на крепкие растворы её солей: в начале происходит переход от красного цвета к жёлтому, а затем в виде маслянистой жидкости частично выделяется тиоугольная кислота (т. пл. -27 °С). Она постепенно разлагается на CS2 и Н2S, но всё же она несравненно устойчивее угольной кислоты. Её кислотные свойства (К1 = 2·10-3, К2 = 7·10-9) также выражены гораздо более сильно.
СИНИЛЬНАЯ КИСЛОТА И ЦИАНИДЫ.
Реакция соединения углерода с азотом сильно эндотермична и частично протекает только при очень высоких температурах. Из простейших азотистых производных углерода наиболее важен цианистый водород (НСN). Он может быть получен из СО и аммиака по реакции:
СО + NH3 + 46 кДж = Н2О + НСN
в присутствии ThO2 (как катализатора), достаточно быстро идущей уже около 500 °С. Цианистый водород представляет собой очень летучую бесцветную жидкость со слабым своеобразным запахом и вкусом (горького миндаля).
С водой НСN смешивается в любых соотношениях, образуя цианистоводородную (синильную) кислоту. Её кислотные свойства выражены крайне слабо, и поэтому она легко выделяется из своих солей (цианидов) действием более сильных кислот.
Синильная кислота применяется главным образом для синтезов органических веществ, а её соли (NaCN, KCN) – при добыче золота. Синильная кислота и её соли чрезвычайно ядовиты. Подобно самому иону СN-,большинство цианидов бесцветно. Производные наиболее активных металлов хорошо растворимы в воде, а менее активных, как правило, малорастворимы.
Цианистый водород может быть получен при 900 °С по уравнению:
2 СН4 + 2 NH3 + 3 O2 = 6 H2O + 2 HCN + 961 кДж
путём пропускания газовой смеси сквозь контактный аппарат с платиновыми сетками. Для лабораторного получения чистой водной НСN (т. пл. -13, т. кип. +26 °С) лучше всего воспользоваться нагреванием растёртой в порошок смеси сухих КСN и КНS. Молекулы цианистого водорода обнаружены в межзвёздной среде.
Обычная синильная кислота содержит смесь молекул Н–СєN (нормальная форма) и Н–NєC (изоформа). Обе формы способны легко переходить друг в друга (путём перескока протона). Поэтому они находятся между собой в динамическом равновесии, положение которого зависит от температуры. При обычных условиях синильная кислота находится почти исключительно (примерно на 99,5%) в виде нормальной формы, а при нагревании равновесие несколько смещается в пользу изоформы. Последняя не была выделена, но имеются указания на ее вероятное образование в некоторых реакциях (например, взаимодействие сухих АgCN и HCl). Органические производные—нитрилы (RСN) и изонитрилы (RNC) — известны для обеих форм синильной кислоты.
Наличие у веществ двух (или более) различных по атомной структуре форм, находящихся в динамическом равновесии друг с другом, говорит о таутомерии данного вещества, а сами формы являются таутомерными его модификациями. В настоящее время установлено, что таутомерия представляет собой довольно распространённое явление. Особенно это относится к таким соединениям у которых в основе таутомерии лежит миграция протона (т.е. внутримолекулярное перемещение водородного ядра от одного атома к другому). Такая миграция обычно сопровождается изменением электронной структуры молекулы.
Молекула НСN линейна. В твёрдом и жидком состоянии цианистый водород ассоциирован за счёт образования водородных связей по схеме ···НСN···HCN···. Частично такая ассоциация сохраняется в парах. При поджигании на воздухе они сгорают фиолетовым пламенем с образованием Н2О, СО2 и N2 (пределы воспламеняемости 6–40% НСN). Сжиганием цианистого водорода в смеси кислорода со фтором по уравнению
2 НСN + O2 + F2 = 2 HF + 2 CO + N2 + 1020 кДж
может быть достигнута температура пламени около 3700 °С.
Токсическое действие синильной кислоты (и цианидов) вызывается, скорее всего, изоформой (НNC) и сводится в основном к параличу дыхания. В организме синильная кислота довольно легко разрушается с образованием безвредных продуктов, поэтому при несмертельных её дозах после периода острого отравления быстро наступает полное выздоровление.
Средством первой помощи при желудочных отравлениях НСN и её солями служит возможно более быстрое возбуждение рвоты (щекотанием нёба или рвотными, например мыльной водой) и приём внутрь 1%-ного раствора Na2S2O3. При отравлении парами НСN полезно вдыхание аммиака. В случае обморока пострадавшего применяется искусственное дыхание. Предельно допустимой концентрацией НСN в воздухе промышленных предприятий считается 3•10-4 мг/л. Хорошим показателем наличия цианистого водорода в воздухе является табачный дым, который в присутствии НСN становится очень горьким. Отравление НСN возможно и через кожу (даже неповреждённую).
Не будучи хорошим растворителем для большинства солей, жидкая синильная кислота сильно ионизирует их растворимую часть. Это связано с её высокой диэлектрической проницаемостью (158 при 0 °С и 107 при 25 °С). Собственная электрическая диссоциация НСN очень невелика: [H+][CN-] =2•10-19. Растворённые в ней НСlO4, H2SO4, и НNO3 ведут себя как слабые электролиты. Это показывает, что тенденция к присоединению протона для молекулы НСN не характерна. Однако выступать в качестве донора она все же может. Так, с VCl4 образуется чёрный твёрдый комплекс (НСN)2VCl4, начинающий разлагаться лишь выше 40 °С.
Кислотные свойства НСN в водном растворе характеризуются значением К = 6·10-10. Как в безводном состоянии, так и в растворе синильная кислота устойчива лишь при одновременном наличии небольших количеств минеральных кислот (или некоторых солей, например СоС2О4), которые являются её стабилизаторами. Хранение НСN без них (а тем более в присутствии следов щелочей) постепенно ведёт к образованию темноокрашенных твёрдых продуктов полимеризации. Процесс этот иногда (при невыясненных ещё условиях) настолько ускоряется, что происходят даже взрывы синильной кислоты.
В водных растворах имеет место также гидролиз по схеме:
HCN + 2 H2O = HCOONH4
с образованием формиата аммония. Обратно, нагреванием этой соли с Р2О5 может быть получен цианистый водород. При хранении водных растворов цианидов последние медленно разлагаются по уравнениям:
KСN + CO2 + H2O = HCN + KHCO3 и
КСN + 2 H2O = NH3 + HCOOK.
С гипохлоритом идёт реакция по уравнению:
2 NaCN + 5 NaOCl + H2O = 5 NaCl + 2 NaHCO3 + N2.
В индивидуальном состоянии из полимеров НСN известны белые кристаллические (НСN)3 (т. пл. 86 °С) и (НСN)4 (т. пл. 284°С). Тример (симметричный триазин) имеет структуру плоского шестичленного кольца из поочерёдно расположенных атомов N и радикалов СН [d(СН) = 109, d(СN) = 132 пм, РNCN = 127°, РСNC = 113°]. Он малоустойчив и легко гидролизуется до HCOONH4. Тетрамер имеет строение NH2(СN)С=С(СN)NН2.
Для иона СN- чрезвычайно характерно вхождение во внутреннюю сферу комплексных соединений. Общим методом получения комплексных цианидов является действие избытка КСN на соли соответствующих металлов. Первоначально выпадающие при этом осадки простых цианидов растворяются затем в избытке осадителя вследствие образования растворимых комплексных цианидов. Реакции идут, например, по схемам:
CrCl3 + 3 КСN = Cr(СN)3Ї+ 3 КCl и Сr(CN)3 + 3 КСl=К3[Cr(СN)6]
Большинство комплексных цианидов хорошо кристаллизуется из растворов. Устойчивость их сильно зависит от природы комплексообразователя и, как правило, велика.
Основным техническим методом получения цианидов является сплавленное цианамида кальция с углём и содой (или поваренной солью). При 800 °С реакция идёт по уравнению:
СаСN2 + C + Na2CO3 + 84 кДж = СаСО3 + 2 NaCN.
Так как СаСО3 практически нерастворим, цианистый натрий может быть извлечён из сплава водой. Чистый NaCN представляет собой бесцветное кристаллическое вещество, в отсутствие воздуха плавящееся без разложения при 564 °С и легкорастворимое в воде. При 1000 °С пар натрийцианида содержит примерно равное число молекул NaCN и (NaCN)2.
При прокаливании смеси поташа и угля в струе аммиака образуется цианистый калий:
К2СО3 + С + 2 NH3 + 276 кДж = 2 КСN + 3 H2O.
Соль эта в отсутствие воздуха плавится при 635 °С и при более высоких температурах испаряется без разложения. В воде она легкорастворима.
Содержащийся в цианидах ион СN-имеет (при свободном вращении) эффективный радиус 192 пм. Из малорастворимых цианидов наиболее важен белый АgCN (ПР = 7·10-15). Для металлов подгрупп Мп, Сr и V простые цианиды нехарактерны, а их комплексные цианиды довольно многочисленны. Типичной особенностью иона СN- при его вхождении во внутреннюю сферу является резкое повышение устойчивости не характерных для элемента-комплексообразователя (в его обычных соединениях) низших степеней окисления.
При нагревании цианистого серебра до 350 °С по реакции:
2 АgCN = (CN)2 + 2 Ag
выделяется дициан (NєC–СєN). Он представляет собой бесцветный ядовитый газ со слабым своеобразным запахом. По ряду химических свойств дициан очень похож на галогены, причём роль атома галогена играет одновалентный радикал СN.
Удобный метод получения дициана основан на реакции:
Hg(CN)2 + HgCl2 = Hg2Cl2 + (CN)2,
которая идёт уже при слабом нагревании смеси сухих солей. Другим удобным методом его получения является проводимая в растворе реакция по уравнению:
4 NaCN + 2 CuSO4 = 2 Na2SO4 + 2 CuCNЇ + (CN)2.
Следы дициана (т. пл. -28, т. кип. -21 °С) всегда содержатся в табачном дыме. Образование его из элементов связано с поглощением тепла (309 кДж) и частично происходит при горении электрической дуги в атмосфере азота.
Молекула дициана линейна (d(CC) = 139, d(CN) = 116 пм). Связь С–С характеризуется энергией 552 кДж/моль. В жидком состоянии дициан ассоциирован и является плохим растворителем для большинства веществ. Будучи подожжён на воздухе, он с большим выделением тепла (1087 кДж/моль) сгорает пурпурным пламенем до СО2 и N2. Сжиганием дициана в кислороде может быть получено пламя с температурой до 4500, а в атмосфере озона — даже до 5000 °С.
В воде дициан хорошо растворим (приблизительно 4:1 по объёму) и постепенно разлагается ею в основном по схеме:
(СN)2 + 4 H2O = (NH4)2C2O4
c образованием оксалата аммония. Нагреванием последнего в присутствии Р2О5 может служить методом получения дициана.
При длительном хранении дициана, действии на него ультрафиолетовых лучей при нагревании выше 500 °С он превращается в твёрдый тёмноокрашенный полимер (“парациан”), который всегда образуется при получении дициана термическим разложением цианидов. Парациан нерастворим в воде, спирте или жидком циане, но растворяется в холодной концентрированной серной кислоте, причём разбавление такого раствора водой сопровождается осаждением парациана. Нагревание его до 860 °С в токе азота ведёт к образованию дициана, а нагревание в токе водорода — НСN, NH3 и свободного углерода.
Известен и другой полимер дициана — гексациан, представляющий собой бесцветные кристаллы (т. пл. 119, т. кип. 262 °С). Строение этого вещества отвечает плоскому шестиугольнику из поочерёдно расположенных атомов N и групп ССN. В присутствии сильно нагретой платины гексациан разлагается с образованием дициана.
Нагревание дициана выше 1000 °С ведёт к его диссоциации по схеме:
С2N2Ы 2 CN.
Радикал СN характеризуется d(CN) = 117 пм и энергией диссоциации 815 кДж/моль. По большинству аналогичных галогенам свойств он располагается между бромом и иодом. Термическая стойкость этого радикала столь велика, что он обнаружен даже в атмосфере Солнца.
При взаимодействии с крепкой соляной кислотой дициан присоединяет две молекулы воды и переходит в оксамид — (СОNH2)2. Последний является белым кристаллическим порошком, нерастворимый в воде. При нагревании он возгоняется с частичным разложением.
Галогениды циана (СlCN, BrCN, ICN) могут быть получены действием соответствующего свободного галогена на водный раствор НСN, Для иода реакция по схеме:
I2 + HCN = ICN + HI
заметно обратима. Хлористый циан представляет собой бесцветный газ (т. пл. -7, т. кип. +13 °С), а BrCN (т. пл. 51, т. кип. 61 °С) и ICN (т. возгонки 140, т. пл. 146 °С под давл.) — летучие кристаллические вещества. Они характеризуются линейной структурой, весьма ядовиты. Пары их уже в самых незначительных концентрациях вызывают сильнейшее слезотечение. Растворимость в воде по ряду Cl-Br-I заметно уменьшается. При хранении галогенцианиды способны полимеризоваться по схеме 3 ГСN = (ГСN)3 с образованием шестичленных колец из поочерёдно расположенных атомов N и групп СГ (особенно легко полимеризуется FCN). Тримеры носят название галогенидных циануров и представляют собой летучие кристаллические вещества. Например, хлористый цианур плавится при 146 и кипит при 190 °С. Взаимодействие его со SbF3 может быть получен фтористый цианур (т. пл. -38, т. кип. +74 °С), термическое разложение которого является лучшим методом получения FCN.
Взаимодействием АgCN с сероуглеродным раствором SCl2 может быть получен цианид серы — S(CN)2. Это легко возгоняющееся бесцветное кристаллическое вещество (т. пл. 61 °С), растворимое в воде и ряде органических жидкостей. Известен и его оранжево-красный полимер, а также аналогичные S(CN)2 цианиды селена и теллура. По схеме:
NH3 + NH2CN = (NH2)2CNH
из цианамида можно получить гуанидин. Структурно соединение это подобно мочевине, в которой атом кислорода замещён на имидную группу. Гуанидин представляет собой бесцветное, очень гигроскопичное кристаллическое вещество (т. пл. 50 °С с разл.). По своей химической функции он является сильным однокислотным основанием и с типичными кислотами образует устойчивые соли. С(NH2)3NO3 (т. пл. 217 °С) находит применение в качестве взрывчатого вещества. В присутствии щелочей гуанидин гидролизуется до мочевины и аммиака.
Взаимодействие дициана со щелочами протекает аналогично подобным же реакциям свободных галогенидов — с одновременным образованием солей синильной и циановой (НNCO) кислот:
(СN)2 + 2 KОН = КСN + KNCO + H2O.
Цианаты могут быть получены также осторожным окислением цианидов, в частности путём сплавления их с оксидом свинца. Цианат калия образуется и при нагревании KСN на воздухе. Соль эта легкорастворима в воде, причём постепенно разлагается ею по схеме:
КNCO + 2 H2O = NH3 + KHCO3.
Термическое разложение цианата калия идёт, в основном по уравнению:
4 KNCO = 2 KCN + K2CO3 + CO + N2.
Цианат серебра бесцветен и малорастворим в воде (ПР = 2•10-7).
Для циановой кислоты (т. пл. -87, т. кип. +25 °С) вероятно следующее равновесие таутомерных форм:
Н-N=C=O Ы NєC-O-H.
При обычных условиях оно смещено влево (тогда как при охлаждении несколько смещается вправо). Взаимодействием НСl с натрийцианамидом при -80 °С было получено 97% НNCO и 3% HOCN, а взаимодействие цианата серебра с SiCl4 в бензоле дало 98% Si(NCO)4 (т. пл. 26, т. кип. 186 °С) и 2% Si(OCN)4 (т. пл. 35, т. кип. 247 °С).
В разбавленном водном растворе циановая кислота (К = 3·10-4) быстро гидролизуется по схеме:
НNCO + H2O = CO2 + NH3
c последующим образованием мочевины:
NH3 + HNCO = CO(NH2)2.
В крепких растворах происходит полимеризация с образованием трёхосновной циануровой кислоты (НNCO)3. Её можно получить нагреванием мочевины или гидролитическим разложением хлористого цианура. Циануровая кислота представляет собой бесцветное кристаллическое вещество, малорастворимое в воде (1:400). Её нагревание ведёт к деполимеризации с получением свободной циановой кислоты.
Тот же элементарный состав, что и циановая, имеет гремучая кислота. Её формула Н-С=N=O с параметрами d(HC) = 103, d(CN) = 116, d(NO) = 121 пм.
Обе кислоты — и циановая и гремучая (соли последней носят название фульминатов) — в свободном состоянии неустойчивы. Из их солей наиболее интересны аммонийцианат (NH4NCO), аргентоцианат (AgNCO), аргентофульминат (AgCNO) и меркуродифульминат [Hg(CNO)2]. Аммонийцианат сыграл большую роль в развитии химии, так как послужил исходным веществом для впервые осуществлённого искусственного синтеза органического вещества (мочевины). Синтез обоих солей серебра дал первое в истории химии указание на существование изомерии (1824 г.). Гремучая ртуть взрывается при ударе и применяется в качестве детонатора. Распад её идёт по схеме:
Hg(CNO)2 = Hg + 2 CO + N2 + 495 кДж.
Пистоны ружейных патронов часто содержат смесь гремучей ртути (25%) с бертолетовой солью (50%) и трёхсернистой сурьмой (25%).
Взаимодействием АgNCO и I2 в ССI4 может быть получен свободный оксоциан — (NСО)2. Он устойчив только при низких температурах и представляет собой бесцветное кристаллическое вещество (т. пл. -12 °С).
Кипячение раствора цианистого калия с серой (или сплавление обоих веществ) сопровождается образованием соли роданистоводородной кислоты (Н-N=C=S) по схеме:
КСN + S = КNCS + 92 кДж.
Свободная НNCS бесцветна и устойчива лишь при очень низких температурах или в разбавленном водном растворе (ниже 5%). В растворе диссоциирована довольно сильно. Большинство её солей (называемых роданидами) бесцветно, хорошо растворимо в воде и при обычных условиях устойчиво. Наиболее распространены соли аммония и калия.
Присоединение серы к солям синильной кислоты лучше всего протекает при действии на них легко отщепляющего серу полисульфида аммония по схеме (для двухсернистого аммония):
KCN + (NH4)2S2 = КNCS + (NH4)2S.
Роданистый аммоний обычно получают взаимодействием (при 110 °С под давлением) крепкого раствора аммиака с сероуглеродом в присутствии гашёной извести по реакции:
2 NH3 + CS2 + Ca(OH)2 = NH4NCS + CaSЇ + 2 H2O.
В противоположность цианидам, соли роданистоводородной кислоты не ядовиты. Ничтожные их количества содержатся в слюне человека.
Свободный роданистый водород может быть получен взаимодействием в вакууме сухих КNCS и KHSO4 c охлаждением выделяющихся паров жидким воздухом. Образующаяся кристаллическая масса плавится при -110 °С. Уже выше -90 °С она начинает полимеризоваться, давая в начале белые, а затем окрашенные твёрдые продукты. Полимер плавится около +3 °С с разложением.
В парообразном состоянии роданистый водород мономолекулярен, причём строение его отвечает формуле Н-N=C=S с параметрами d(HN) = 99, d(NC) = 122, d(CS) = 156 пм, РHNC = 135°.
Взаимодействие HNCS с сероводородом ведёт к образованию СS2 и NH3. При нагревании HNCS c не очень крепкой серной кислотой реакция идёт в основном по схеме:
HNCS + H2O + H2SO4 = NH4HSO4 + COS.
Процесс этот может быть использован для получения тиооксида углерода. Сильные окислители (Н2О2, KMnO4 и т. п.) переводят HNCS в HCN и Н2SO4. Кислотные свойства роданистоводородной кислоты характеризуются значением К = 0,5 (т.е. она гораздо сильнее циановой). Кислота эта содержится в соке лука.
Прямое определение строения роданистого водорода, приведшее к современной формуле Н-N=C=S, было выполнено сравнительно недавно.
Смешивание КNCS c водой (3:2 по массе) сопровождается понижением температуры образующегося раствора приблизительно на 30 °С, что используется иногда в составах против обледенения. Некоторые роданиды (особенно Li) имеют сильно выраженную склонность к образованию пересыщенных растворов. При нагревании KNCS (т. пл. 177 °С) в отсутствие кислорода примерно до 400 °С расплав синеет вследствие частичной термической диссоциации роданида на КCN и S с последующим образованием коллоидного раствора серы (образованной молекулами S4) в избытке KNCS.
Интересен образующийся при растворении MnCO3 в НNCS роданид марганца. В противоположность розово-красной окраске почти всех остальных производных Мn2+, безводный Мn(NCS)2 жёлтый, а его кристаллогидрат Мn(NCS)2·4Н2О ярко-зелёный. При растворении в воде первоначально образуется зелёный раствор, который становится розовым лишь после достаточного разбавления. Из нерастворимых роданидов наибольшее значение имеет белое роданистое серебро — AgSCN (ПР = 1·10-13). Кристаллы этой соли слагаются из цепей типа .···АgSCN···AgSCN···с параметрами d(AgS) = 243, d(AgN) = 222, d(CN) = 119, d(CS) = 164 пм. Цепи изогнуты у атомов S и Аg (РАgSC = 104°, РNAgS = 165°) и имеют поэтому форму зигзага. Взаимодействием AgSCN с SO2CI2 был получен нестойкий SO2(SCN)2.
Действием на AgSCN брома (в сероуглеродном растворе) по реакции:
2 AgSCN + Br2 = 2 AgBr + (SCN)2
может быть получен свободный родан (SCN)2, строение молекулы которого отвечает формуле NCS-SCN. Он представляет собой устойчивые только при низких температурах бесцветные кристаллы (т. пл. -2 °С). В воде родан хорошо растворим, но быстро разлагается по уравнению:
3 (SCN)2 + 4 Н2O = 5 HCNS+ Н2SO4 + HCN.
Подобно свободным галогенидам, родан непосредственно соединяется с некоторыми металлами, образуя роданиды. Сродство к электрону радикала NCS оценивается в 209 кДж/моль. Окислительная функция выражена у родана слабее, чем у брома, но сильнее, чем у иода. При хранении он легко переходит в красный полимер (SCN)п. Для родана известны продукты присоединения типов (SCN)2·НГ (где Г— Сl, Вr) и (SCN)2·H2Э (где Э — O, S).
Термическое разложение роданида ртути идёт по уравнению:
2 Hg(SCN)2 = 2 HgS + CS2 + C3N4
c образованием нормального нитрида углерода (С3N4). В индивидуальном состоянии его удобнее получать термическим разложением цианистой серы:
2 S(CN)2 = CS2 + C3N4.
Нитрид углерода представляет собой чрезвычайно объёмистую аморфную массу жёлтого цвета, сильно поглощающую влагу, но нерастворимую ни в воде, ни в каком-либо другом растворителе. При нагревании до температуры красного каления он разлагается на циан и свободный азот.
По элементарному составу к роданистоводородной кислоте близок рубеановый водород (С2S2N2H4), образующийся по уравнению:
2 НСNS + 2 H2O = C2S2N2H4 + H2
Это красно-оранжевое вещество, разлагающееся при 170 °С. В воде он растворим сравнительно мало (0,02 моль/л), причём разлагается ею с образованием щавелевой кислоты, аммиака и сероводорода. Обладая весьма слабо выраженными кислотными свойствами (К1 = 3·10-10), рубеановый водород даёт с катионами ряда металлов труднорастворимые и характерно окрашенные соединения. В частности он является очень чувствительным реактивом на медь.
ГАЛОГЕНИДЫ УГЛЕРОДА.
Простейшие галогениды углерода отвечают формуле СГ4. Взаимодействие элементов может быть получено только фтористое производное, а остальные получают косвенным путём.
Наиболее практически важен четырёххлористый углерод (ССl4). Он представляет собой тяжёлую бесцветную жидкость со слабым характерным запахом. С химической стороны он характеризуется главным образом своей инертностью. Так, при обычных условиях ССl4 не вступает во взаимодействие ни с кислотами, ни со щелочами.
Четырёххлористый углерод прекрасно растворяет жиры, масла, смолы, многие краски и т. п. и может поэтому служить хорошим средством для вывода пятен. Так как он не горюч, при работе с ним устраняется пожарная опасность, что даёт ССl4 значительное преимущество перед более дешёвым растворителем перечисленных выше веществ — сероуглеродом.
Теплоты образования из элементов газообразных галогенидов СГ4 очень сильно зависят от природы галогена (кДж/моль): 932 (F), 134 (СI), -84 (Br), -305 (I). Два последних соединения являются эндотермическими.
ССl4 (т. пл. -23, т. кип. 77 °С) получают действием хлора на СS2. При нагревании до 60 °С в присутствии катализатора (например, FeS) реакция идёт по уравнению:
СS2 + 2 Cl2 = CCl4 + 2 S + 226 кДж.
Несмотря на химическую инертность четырёххлористого углерода, некоторые металлы (например АI, Fe) заметно разъедаются им. В их присутствии ССI4 уже при обычных температурах постепенно разлагается водой по схеме:
ССl4 + 2 H2O = CO2 + 4 HCl.
Его собственная растворимость в воде очень мала (5·10-3 моль/л). На негорючести и тяжести паров четырёххлористого углерода основано его применение в некоторых системах огнетушителей. Он ядовит и предельно допустимым содержанием его паров в воздухе промышленных предприятий считается 0,02 мг/л. Ежегодная мировая выработка ССl4 составляет около 200 тыс. т.
Аналогичные четырёххлористому углероду производные других галогенов обычно получают обменным разложением ССl4 при нагревании соответственно с АgF, AIBr3 или АII3. Четырёхфтористый углерод может быть получен также непосредственным взаимодействием углерода со фтором, энергично протекающим уже при обычных условиях (тогда как кусковой графит устойчив по отношению ко фтору почти до 400 °С). Молекулы галогенидов СГ4 представляют собой правильные тетраэдры с расстоянием С-Г, равным: 132 (СF), 177 (CCl), 194 (CBr), 215 пм (CI).
Четырёхфтористый углерод газообразен (т. пл. -184, т. кип. -128 °С), а СВr4 (т. пл. 90, т. кип. 187 °С) и СI4 (т. пл. 171 °С с разложением на 2 I2 и С2I4) представляют собой твёрдые вещества. В противоположность остальным галогенидам СГ4, которые бесцветны, СI4 имеет тёмно-красную окраску. Он может быть возогнан в вакууме (ниже 100 °С), с жидким аммиаком образует нестойкий жёлтый аммиакат СI4·2NH3, а в присутствии амида калия реагирует по схеме:
СI4 + KNH2 + NH3 = KI + CHI3 + N2H4.
По химическим свойствам все рассматриваемые галогениды в общем похожи на ССl4, а устойчивость их уменьшается по ряду F-Cl-Br-I. Выше -170 °С жидкий СF4 в любых соотношениях смешивается с жидким озоном (и может быть использован для его разбавления).
Диссоциация СF4 на фтор и имеющий пирамидальное строение трифторметильный радикал (СF3) требует затраты 581 кДж/моль. Известно очень много производных этого радикала. Обычно их получают при помощи СF3I (т. кип. -22 °С), распад которого на I и CF3 протекает с очень небольшой энергией активации (8 кДж/моль).
МЕТАН.
В обычных условиях непосредственное взаимодействие углерода (аморфного) и водорода с образованием метана (СН4) по реакции
С + 2 Н2Ы СН4 + 75 кДж
практически не происходит. При нагревании и в присутствии катализатора (мелкораздробленный Ni) устанавливается равновесие, положение которого сильно зависит от температуры (рис. 8). Помимо этого синтетического пути, метан может быть получен рядом других методов из более сложных соединений углерода. В природе он постоянно образуется при разложении органических веществ без доступа воздуха (например, в болотах). Он часто содержится в природных газах и обычно входит в состав искусственно получаемого светильного газа.
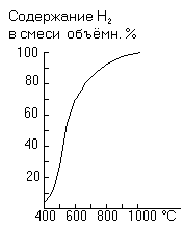
Рис. 8. Равновесие синтеза метана.
Метан является простейшим представителем многочисленных соединений углерода с водородом, называемых углеводородами и изучаемых в органической химии. Сам он представляет собой бесцветный и не имеющий запаха газ, малорастворимый в воде. С химической стороны метан характеризуется своей большой инертностью, на него не действуют ни щёлочи, ни кислоты. С кислородом он в обычных условиях не реагирует, но при поджигании сгорает по реакции:
СН4 + 2 О2 = СО2 + 2 Н2О.
Горение метана сопровождается очень большим выделением тепла (803 кДж/моль).
Метан является основой атмосфер тяжёлых планет (Юпитера, Сатурна). Следы его (1,4·10-4 объёмн. %) всегда содержатся в земной атмосфере. Интересно, что повышение содержания метана привлекает комаров.
Большие количества метана (часто свыше 90%) содержат многие скопившиеся в подземных пустотах природные газы. Такие газы являются очень хорошим топливом, 1 м3 которого даёт при сгорании 33-38 тыс. кДж. Вместе с тем они служат основным сырьём для промышленного получения водорода. Обычно применяемый при этом метод основывается на неполном окислении метана по уравнению:
2 СН4 + О2 = 2 СО + 4 Н2 + 71 кДж.
Получаемый газ подвергается затем вторичной обработке водяным паром.
На территории СНГ известны многочисленные месторождения богатых метаном природных газов (Саратов, Ставрополь, Бухара и др.). Благодаря высокой калорийности и удобству транспортировки (по трубопроводам), такой газ с каждым годом занимает всё более важное место в общем топливном балансе страны.
Горение смеси метана (и других горючих газов) с воздухом идёт только в том случае, если процентный состав смеси не выходит из некоторых определённых границ. Так, пределы воспламеняемости (в процентах горючего газа по объёму) составляют 5-15 для метана, 12-74 для СО и 4-75 для Н2. Данные эти относятся к обычному давлению и являются ориентировочными, так как помимо давления пределы воспламеняемости зависят и от других условий.
При местном нагревании смеси, имеющей подходящий для воспламенения состав, горение почти мгновенно распространяется по всему её объёму и происходит взрыв. В качестве горючего материала подобных взрывчатых смесей с воздухом способны фигурировать не только газы или пары, но и пыль различных горючих веществ (угля, муки, сахара и т. п.). Этим могут быть обусловлены происходящие иногда взрывы на элеваторах, сахарных заводах и т. п.
Чтобы поджечь газовую смесь, необходимо хотя бы в одном месте нагреть её до некоторой минимальной температуры, которая носит название температуры воспламенения. Последняя зависит не только от природы реагирующих газов, процентного состава смеси и давления, но и от способа зажигания и некоторых других условий. Поэтому она не является постоянной величиной, а колеблется в некоторых пределах. Так, смесь метана с воздухом воспламеняется под обычным давлением при 650-750, СО — при 610-660, водорода — при 510-590 °С.
Само существование определённых температур воспламенения тесно связано с энергиями активации соответствующих реакций. При этом, способные к химическому взаимодействию составные части газовой смеси реагируют друг с другом и при более низких температурах, чем то отвечает появлению пламени, т.е. быстро протекающей реакции, сопровождающейся выделением тепла и света. Однако взаимодействие при подобных условиях происходит только между отдельными достаточно активными молекулами и даёт поэтому лишь сравнительно небольшое количество тепла, которое быстро рассеивается вследствие теплопроводности, лучеиспускания и т. д.
По мере повышения температуры места нагрева число активных молекул около него растёт, и выделение тепла в результате их взаимодействия соответственно увеличивается. При определённых температурных условиях (соответствующих температуре воспламенения) вблизи места нагрева создаётся такое положение, когда за единицу времени тепла больше выделяется, чем рассеивается. Это приводит к нагреву до температуры воспламенения соседних участков системы, от которых подобным же образом массовая активация молекул распространяется на дальнейшие и т. д. Результатом является резкое увеличение скорости процесса во всём реакционном пространстве, внешне выражающееся в появлении пламени.
Характер горения в той или иной системе определяется её внутренней структурой. Если составные части системы хорошо перемешаны друг с другом, пламя быстро распространяется на весь её объём и происходит взрыв. Напротив, если реагирующие газы смешиваются лишь в самый момент реакции (как в обычных горелках), горение протекает только в местах их соприкосновения и получается спокойное пламя.
Горение жидкостей и твёрдых тел может происходить лишь на поверхности их соприкосновения с воздухом, поэтому оно обычно протекает спокойно. Кроме температуры воспламенения (определяемой началом горения всей поверхности), горючесть жидкостей часто характеризуют температурой вспышки. Под последней понимается та минимальная температура жидкости, при которой поднесение пламени вызывает вспышку её паров (но сама она не загорается). Например, по стандарту температура вспышки продажных сортов керосина не должна быть ниже 28 °С. Воспламенение керосина происходит около 300 °С.
КАРБИДЫ.
С металлами углерод вступает во взаимодействие лишь при высоких температурах. Из образующихся соединений (называемых карбидами) наибольшее практическое значение имеет карбид кальция (СаС2). Весьма важны также производные W (W2C и WC).
Большинство карбидов удобнее получать прокаливанием с углём оксидов металлов. При высоких температурах происходит восстановление оксидов, причём металл соединяется с углеродом. Прокаливанием в электрической печи смеси угля с оксидом кальция получают и карбид кальция:
СаО + 3 С + 464 кДж = СО + СаС2.
Технический продукт окрашен в серый цвет примесью свободного углерода. Чистый СаС2 представляет собой бесцветные кристаллы, образованные ионами Са2+ и С22-. Получение каждой тонны карбида кальция требует затраты 3 тыс. кВт·ч. Его ежегодная мировая выработка составляет около 5 млн. т.
Карбиды представляют собой твёрдые, в чистом состоянии хорошо кристаллизующиеся вещества. Они нелетучи и не растворимы ни в одном из известных растворителей. В связи с этим истинные молекулярные веса карбидов неизвестны, и для них приходится довольствоваться простейшими формулами.
Последние в одних случаях соответствуют обычным валентностям углерода и соединяющегося с ним металла, в других — уже сами по себе указывают на сложность молекулярной структуры карбида. В этом отношении, а также и по ряду свойств (устойчивости большинства при нагревании и т. д.) карбиды очень похожи на нитриды.
По отношению к воде и разбавленным кислотам карбиды распадаются на две большие группы: разлагаемые этими веществами и не разлагаемые ими. Карбиды первой группы в зависимости от химической природы летучих продуктов их разложения можно в свою очередь подразделить на: а) ацетилениды; б) метаниды и в) дающие смесь различных продуктов.
Карбиды первого типа следует рассматривать как продукты замещения водорода ацетилена. Их образуют главным образом наиболее активные металлы. Общая формула карбидов этой подгруппы имеет вид М2С2 для одновалентного металла, МС2 — для двухвалентного и М2С6 — для трёхвалентного.
Подобным же образом карбиды второго типа следует рассматривать как продукты замещения на металл водородов метана. Известны они только для бериллия и алюминия, причём в обоих случаях простейшие формулы (Ве2С и Аl4C3) отвечают обычным валентностям элементов. При действии горячей воды или разбавленных кислот оба карбида разлагаются с выделением чистого метана, например, по схеме:
Al4C3 + 12 H2O = 4 Al(OH)3 + 3 CH4.
Примером карбидов третьего типа, дающих при разложении смесь различных продуктов, может служить Mn3C, который реагирует с водой преимущественно по уравнению:
Mn3C + 6 H2O = 3 Mn(OH)2 + CH4 + H2.
Одновременно образуются также и другие газообразные углеводороды.
Неразлагаемые разбавленными кислотами карбиды обычно очень устойчивы также по отношению к другим химическим воздействиям и нагреванию. Все они могут быть, однако, разрушены сплавлением со щелочами при доступе воздуха (например, по реакции:
2 WC + 8 NaOH + 5 O2 = 2 Na2WO4 + 2 Na2CO3 + 4 H2O).
Некоторые карбиды этой группы проводят электрический ток. Из производных уже рассматривавшихся металлов сюда относятся: Mn23C6, Mn3C, Mn7C3, TcC, Cr3C2, Cr7C3, Cr23C6, MoC, Mo2C, WC, W2C, VC, NbC, Nb2C, TaC, Ta2C. Многие соединения этого типа принадлежат к наиболее тугоплавким из всех известных веществ. Примерами могут служить WC (т. пл. 2600 °С с разл.), W2C (2700), VC (2800), NbC (3500), TaC (3900 °С). Сплавы на основе карбида хрома весьма стойки к коррозии и износу. Сцементированный никелем карбид тантала под названием “рамет” находит применение в качестве сверхтвёрдого сплава, а карбиды Nb и Ta — в ракетной технике.
С водой (даже со следами) карбид кальция энергично реагирует, образуя ацетилен (Н-СєС-Н) по уравнению:
СаС2 + 2 Н2О = Са(ОН)2 + С2Н2 + 125 кДж.
Получаемый из технического СаС2 ацетилен имеет неприятный запах вследствие наличия в нём ряда примесей (NH3, PH3, H2S и др.). В чистом виде он представляет собой бесцветный газ со слабым характерным запахом, довольно хорошо растворимый в воде.
Ацетилен служит исходным продуктом для синтеза очень многих сложных органических соединений. Эта область его использования и является самой обширной. Другое важное применение ацетилена основано на протекающей с большим выделением тепла реакции его сгорания:
2 С2Н2 + 5 О2 = 4 СО2 + 2 Н2О + 2510 кДж.
Развивающейся при горении ацетилена (в смеси с кислородом) высокой температурой (около 3000 °С) пользуются для “автогенной” сварки и резки металлов. На воздухе ацетилен горит белым пламенем, сильно коптящим вследствие неполного сгорания углерода.
Как и в случае синильной кислоты, для ацетилена (т. возг. -84, т. пл. -81 °С под давл.) возможна таутомерия с образованием двух форм: Н-СєС-Н (ацетилен) и Н2С=С (изоацетилен). При обычных условиях равновесие практически нацело смещено в сторону нормальной формы, а при нагревании несколько смещается, по-видимому, в сторону изоформы. Критическая температура ацетилена +35 °С.
Образование ацетилена из элементов идёт лишь выше 2000 °С и сопровождается поглощением тепла (226 кДж/моль). Будучи сильно эндотермичным соединением, ацетилен способен разлагаться со взрывом. В газообразном состоянии такой распад при обычных условиях не происходит, но под повышенным давлением, и особенно в жидком или твёрдом состоянии, может произойти от самых ничтожных воздействий (сотрясения и т. п.). Растворимость ацетилена в воде (1:1 по объёму при обычных условиях) значительно меньше, чем в различных органических растворителях. Охлаждением насыщенного водного раствора может быть получен кристаллогидрат С2Н2·6Н2О.
Водороды ацетилена имеют очень слабо выраженный кислотный характер (К1 = 10-14). Для него известны соли некоторых металлов (ацетилениды), как правило, взрывчатые. Сравнительно устойчивые бесцветные соли натрия — средняя (Na2C2) и кислая (NaHC2) — могут быть получены действием ацетилена на раствор NaNH2 в жидком аммиаке. Известны и некоторые комплексные соединения, содержащие ионы [CєCH]- во внутренней сфере. Примерами могут служить розовый К2[Mn(C2H)4] и оранжевый К3[Cr(C2H)6]. Оба они очень неустойчивы.
В молекуле диацетилена — НСєС-СєСН — центральная связь С-С имеет длину 138 пм при неизменности по сравнению с ацетиленом длины связей СєС. Это бесцветный газ (т. пл. -35, т. кип. +10 °С), легко полимеризующийся. Известен и триацетилен.
Дальнейшей полимеризацией ацетилена может быть получен полиацетилен (полиин), обладающий полупроводниковыми свойствами.
Для автогенной сварки и резки металлов пользуются специальной горелкой, содержащей три вставленные друг в друга трубки. Ацетилен входит по средней трубке, кислород — по обеим крайним, благодаря чему достигается лучшее перемешивание газов. Кислород поступает из содержащих его баллонов, а ацетилен или получают на месте работы, или выделяют из раствора его в ацетоне. Под давлением 12 атм 1 объём ацетона растворяет 300 объёмов С2Н2, под обычным давлением — только 25. Поэтому при открытии крана у баллона с таким раствором из него выделяется ток С2Н2. Содержащие его баллоны имеют белую окраску с красной надписью “Ацетилен”.
Образующиеся при неполном сгорании С2Н2 твёрдые частички углерода, сильно накаляясь, обуславливают яркое свечение пламени, что делает возможным использование ацетилена для освещения. Применением специальных горелок с усиленным потоком воздуха удаётся добиться одновременного сочетания яркого свечения и отсутствия копоти: сильно накаливающиеся во внутренней зоне пламени частички углерода затем сполна сгорают во внешней зоне. Газы, не образующие при сгорании твёрдых частиц (например, Н2), в противоположность ацетилену, дают почти несветящееся пламя. Так как в пламени обычно применяемых горючих веществ (соединений С с Н и отчасти О) твёрдые частички могут образовываться за счёт неполного сгорания только углерода, пламя газов и паров жидкостей бывает при одних и тех же условиях тем более коптящим, чем больше относительное содержание в молекулах горящего вещества углерода и меньше кислорода и водорода. Например, спирт (С2Н5ОН) горит некоптящим пламенем, а скипидар (С10Н16) — сильно коптящим. Яркость пламени зависит и от степени накаливания этих твёрдых частиц, т.е. от развивающейся при горении температуры.
Углерод во всех своих наиболее устойчивых соединениях четырёхвалентен. Единственным исключением является оксид углерода (II), но и он, как уже отмечалось, склонен к реакциям присоединения, сопровождающимся переходом углерода в четырёхвалентное состояние. Кроме СО известно лишь очень немного производных углерода с валентностью, иной, чем четыре (а именно 2 и 3), но подобные соединения при обычных условиях малоустойчивы.
Круговорот углерода в природе.
История углерода в далёком прошлом нашей планеты ещё не ясна. Согласно разработанной в 1944 году О. Ю. Шмидтом и ныне почти общепринятой космогонической теории, Земля формировалась более 5 миллиардов лет тому назад не из раскалённой массы газов, как полагали ранее, а из пылевидных частиц холодного космического вещества. Относительно происхождения исходного гигантского облака такого вещества, его температуры и химического состава пока нет единого мнения.
Первоначально предполагалось, что облако космического вещества было захвачено Солнцем на части его пути вокруг центра Галактики (проходимого со скоростью 220 км/с за время около 200 млн. лет). Затем было выдвинуто предположение об этом облаке как остатке материала от формирования самого Солнца. Наконец, возможно (и даже наиболее вероятно) предположение о выбросе материала облака из недр уже сформировавшегося Солнца.
Пылевидные частицы мирового пространства находятся в условиях высокого вакуума. Вдали от звёзд они имеют равновесную температуру около -270 °С, но по мере приближения к источнику лучеиспускания эта температура повышается. Абсолютно чёрное тело (т.е. тело, полностью поглощающее все падающие на него лучи) на расстоянии Земли от Солнца было бы нагрето приблизительно до +4 °С. Средняя равновесная температура реальных пылинок должна лежать где-то между -270 и +4 °С.
Химический состав космического пылевого облака зависит и от его происхождения (включая время, прошедшее с момента возникновения), и от конечной равновесной температуры. Ни то, ни другое точно не установлено, поэтому намечать этот состав можно лишь предположительно. Скорее всего, он был близок к составу метеоритов. Несомненно, что исходное пылевидное облако содержало (в замороженном состоянии) также и гораздо более летучие вещества.
Стяжение отдельных частиц холодной космической пыли в компактную массу планеты сопровождалось повышением температуры. Дальнейшее разогревание уже сформировавшейся Земли последовало за счёт распада вошедших в её состав радиоактивных элементов. В результате внутренние слои нашей планеты нагревались по крайней мере до 2000 °С. Это сопровождалось интенсивной вулканической деятельностью, в результате которой недра Земли извергли колоссальные количества различных газов и паров (причём главная их масса приходилась на водяной пар). Затем, по мере уменьшения запасов радиоактивных элементов, наступило постепенное охлаждение Земли до её современного состояния.
Существуют две крайние точки зрения на максимально достигавшуюся в прошлом температуру земной поверхности. Согласно одной из них, температура эта превышала 1000 °С. Выносимый тогда из земных недр водяной пар конденсировался лишь после достаточного охлаждения Земли. Согласно другой точке зрения, температура земной поверхности никогда не превышала 100 °С. При этих условиях жидкая вода имелась на поверхности нашей планеты с гораздо более далёких времён.
Независимо от признания “горячего” или “холодного” прошлого земной поверхности, основная масса её вод должна была в конечном счёте происходить из космического льда. Эта мысль была впервые высказана Аристотелем.
В обоих случаях основным веществом атмосферы над первичной земной поверхностью должен был быть водяной пар. Следующее за ним место среди извергаемых недрами Земли газов и паров занимал по количеству углекислый газ. Древняя атмосфера содержала углерод главным образом в виде углекислого газа.
Относительно состава первичной атмосферы Земли имеются две точки зрения. Согласно одной из них, древняя атмосфера слагалась в основном из водяного пара, углекислого газа и свободного азота, тогда как другие газы (СО, СН4, NH3, H2S и др.) содержались в качестве примесей. Согласно другой точке зрения, первичная атмосфера имела восстановительный характер: помимо водяного пара, она состояла главным образом из водорода, метана и аммиака. Под действием солнечного излучения водяной пар разлагался по схеме:
Н2О + hn = H2 + О,
причём водород уходил в верхние слои атмосферы и постепенно терялся Землёй, тогда как кислород расходовался на окисление метана до СО и затем до СО2, а аммиака — до N2. Атмосфера, состоящая в основном из азота, углекислого газа и водяного пара, является вторичной.
Так как фотохимическое разложение водяного пара не прекращалось, в дальнейшем атмосфера начала обогащаться свободным кислородом. Однако до появления растительности такое обогащение шло весьма медленно.
Голая поверхность первичной земной коры не создавала благоприятных условий для возникновения на ней органической жизни. Не было этих условий и в водах первичного океана. Потребовалось много миллионов лет совместной работы различных природных факторов (деятельности вулканов, солнечных лучей, дождя, ветра и др.) для того, чтобы в результате разрушения (“выветривания”) горных пород поверхность земли покрывалась слоем почвы, а воды океана обогатились разнообразными солями. Видную роль в этом процессе разрушения горных пород играл углекислый газ, переводящий металлы первичных минералов в средние и затем в кислые углекислые соли, которые вымывались водой и постепенно накапливались в океане. На данном этапе истории Земли химические взаимодействия СО2 шли, таким образом, исключительно по пути неорганических реакций разрушения первичных минералов земной коры.
Органическая жизнь возникла на Земле более трёх миллиардов лет тому назад, т.е. в период катархея. Мы пока ещё не знаем, как осуществлялся в природе скачкообразный переход от неорганизованной материи к более высокой форме её развития — простейшему живому веществу. Несомненно, однако, что ему предшествовал длительный “подготовительный” период. Условия, при которых происходили эти изменения, сильно отличались от современных. В частности, температура земной поверхности была тогда значительно выше, а атмосфера если и содержала свободный кислород, то лишь в незначительных количествах.
Существует предположение, что главным исходным материалом для построения живого вещества служили углеводороды, возникшие за счёт взаимодействия воды с карбидами металлов. Такое взаимодействие становится возможным при разрывах твёрдой земной коры в процессе её геологического переформирования. Одновременно с углеводородами, за счёт разложения водой нитридов, мог выделяться аммиак, азот которого использовался затем при образовании белковых молекул.
Прямыми опытами было показано, что под действием ультрафиолетовых лучей (или электрических разрядов) на смеси водяного пара с метаном, аммиаком и водородом образуется ряд органических веществ, в том числе различных аминокислот. В отдельные эпохи, когда ещё не существовало защищающего Землю от “жёсткого” солнечного излучения озонового слоя, условия для протекания такого фотохимического синтеза были весьма благоприятны. Так как аминокислоты являются основой белковых тел, первичное возникновение жизни могло быть связано непосредственно с подобными процессами.
Интересны опыты по выяснению возможности возникновения первичного живого вещества под действием только высоких температур. Сначала метан пропускался сквозь раствор аммиака и затем сквозь нагретую до 1000 °С кварцевую трубку, заполненную различными минеральными веществами (кварцем, силикагелем, оксидом алюминия и др.). Полученный продукт содержал 18 аминокислот, имеющихся в белках. Его наносили на нагретый до 170 °С кусок лавы и время от времени орошала дистиллированной водой (имитация дождя). Через несколько часов такого режима на поверхности лавы была обнаружена обширная микроструктура, состоящая из большого числа сферических частиц, образованных связавшимися в цепи аминокислотами.
Исходя из этих результатов можно думать, что первичные агрегаты аминокислот возникали на склонах вулканов. Затем они смывались дождями и уносились в океан, который представлял собой в те времена как бы очень разбавленный “бульон” из простейших соединений. Там эти агрегаты находили благоприятные условия для дальнейшего превращения в простейшее живое вещество.
Обнаружение органических веществ (аминокислот и др.) в некоторых метеоритах указывает на принципиальную возможность зарождения жизни и вне Земли. Однако ни на одном из известных небесных тел не существовало условий (океана с его “бульоном”) для практической реализации такой возможности.
“Жизнь — это способ существования белковых тел, существенным моментом которого является постоянный обмен веществ с окружающей их внешней природой” (Ф. Энгельс). Колыбелью жизни был океан. В нём первично формировались те простейшие комочки живой материи, дальнейшее развитие которых привело к возникновению всего многообразия органического мира.
Ещё миллиард лет тому назад в океане были широко распространены водоросли и имелись представители простейших животных (губки, членистоногие). Лишь впоследствии (около 500 миллионов лет тому назад) жизнь частично перешла и на сушу, где тёплая, влажная, богатая углекислым газом и бедная кислородом атмосфера особенно благоприятствовала развитию растительных форм. В результате 400 миллионов лет тому назад, когда представители животного мира на суше ещё почти отсутствовали, она уже была покрыта богатой растительностью.
Сильное развитие растительности и в океане, и на суше привело к изменению химического состава атмосферы. Постоянно извлекая из неё необходимый им для построения тканей углекислый газ, растения возвращали обратно кислород. Кроме того, значительные количества углекислого газа продолжали тратиться на разрушение горных пород, поэтому содержание его в атмосфере постепенно уменьшалось, в связи с чем развитие на Земле растительности, достигшее своего максимума около 300 миллионов лет тому назад, пошло затем несколько на убыль.
Сильно эндотермический (порядка 468 кДж на моль СО2) процесс усвоения углекислого газа растениями с образованием углеводов может быть суммарно выражен общей схемой
n CO2 + m H2O = Cn(H2O)m + n O2
и осуществляется за счёт энергии солнечных лучей (110 000 млрд. кДж/с для всей земной поверхности). Значение света для развития зелёных растений было известно уже Аристотелю: “Те части растений в которых влажное не смешивается с солнечными лучами, остаются белыми”,— писал он. К. А. Тимирязев (1843-1920) установил, что процесс фотосинтеза протекает под воздействием содержащегося в зелёных частях растений сложного органического вещества — хлорофилла. Коэффициент использования энергии солнечного света при фотосинтезе невелик (в среднем порядка 2%).
Зелёные растения ежегодно усваивают около 550 млрд. т. углекислого газа и выделяют около 400 млрд. т. кислорода. При этом образуется около 380 млрд. т. биомассы. По другим оценкам, ежегодная общая продукция фотосинтеза составляет 85 млрд. т. органического вещества, что соответствует усвоению лишь 150 млрд. т. углекислого газа и выделению 110 млрд. т. кислорода. Соотношение растительной и животной биомасс на всём земном шаре оценивается как 2200:1.
Имея в виду, что после отмирания растительных организмов останки их подвергаются тлению, при котором углерод возвращается атмосфере в виде СО2, в конечном счёте в атмосфере должно было бы установиться определённое равновесное распределение углерода между растительным покровом и атмосферой. Однако этому мешали мощные сдвиги земной коры, зачастую погребавшие под слоями горных пород громадные растительные массивы. Подвергаясь на протяжении миллионов лет разложению под давлением и без доступа кислорода, эти растительные останки переходили во всё более богатые углеродом соединения с образованием в конечном счёте различных ископаемых углей, являющихся ценным наследством, дошедшим до нас от минувших геологических эпох. Содержащийся в них углерод уже не возвращался атмосфере и таким образом выводился из круговорота.
Основными составными частями древесины (не только деревьев, но и трав, мхов и т. п.) является клетчатка [(C6H10O5)x] и лигнин — органическое вещество ещё не установленного строения, более богатое углеродом, чем клетчатка. При разложении отмерших растительных организмов без доступа воздуха (на дне болот, под слоями горных пород) из них выделяются летучие продукты распада, а остаток постепенно обогащается углеродом. Это соответствующим образом сказывается на химическом составе и теплотворной способности продукта разложения, который, в зависимости от его особенностей, называют торфом, бурым углем, каменным углем или антрацитом.
Торф является сравнительно молодым продуктом и сохраняет структуру тех растительных волокон (чаще всего мхов), из которых он образовался. Хотя возраст бурого угля исчисляется уже миллионами лет, на нём легко заметить структуру исходных древесных пород. На более старых каменных углях распознать эту структуру можно лишь в исключительных случаях. Наконец, образовавшиеся из растительности ещё более древних эпох антрациты представляют собой серо-черную плотную массу, на которой какие-либо следы растительной структуры уже совершенно незаметны. Переходной формой от антрацита к графиту является шунгит.
Ископаемые угли представляют собой один из важнейших видов промышленного и бытового топлива. Значительные его количества расходуются для выработки необходимого металлургии кокса. Последний получают сильным нагреванием каменного угля без доступа воздуха. В результате из угля выделяются различные летучие продукты, а в печах остаётся серо-чёрная спёкшаяся масса кокса, выход которого составляет 60-70% от массы взятого угля. Ввиду предварительного удаления летучих веществ кокс сгорает почти без пламени, что делает его особенно пригодным для выплавки металлов из руд. Теплотворная способность кокса равна приблизительно 33500 кДж/кг.
Важными побочными продуктами коксования являются каменноугольная смола (служащая исходным продуктом для получения ряда органических веществ), аммиак и коксовый газ. В состав последнего входит (по объёму) приблизительно 60% Н2, 25 — СН4, 2 — других углеводородов, 5 — СО, 2 — СО2 и 5-6% N2. Благодаря большому содержанию Н2. коксовый газ является хорошим исходным продуктом для получения водорода. С этой целью газовую смесь подвергают сильному охлаждению, причём все её составные части, кроме Н2, сжижаются, и водород может быть поэтому легко отделён.
C коксованием весьма сходен процесс получения из каменного угля светильного газа. Процесс проводят при более низкой температуре, чем коксование, поэтому образующийся газ содержит относительно больше углеводородов, чем коксовый. В состав его входит обычно около 50% Н2, 30 –СН4, 4 – других углеводородов, 9 – СО, 2 – СО2 и 4-5% N2. Ввиду значительного содержания СО светильный газ весьма ядовит. При сжигании газа указанного состава выделяется 23000 кДж/м3. Из тонны каменного угля получается приблизительно 300 м3 светильного газа, 50 л смолы и 3 кг аммиака. Подобным продуктом газификации угля является также кокс. В связи с расширением добычи природного горючего газа светильное производство теряет своё прежнее значение. Однако в будущем оно, вероятно, вновь возрастёт.
При сжигании светильного (или природного) газа в обычных газовых горелках “несветящееся” пламя слагается из трёх конусов. Внутренний конус образован струёй смешанного с воздухом газа, и горения в нём вовсе не происходит. В следующем конусе имеется избыток горючего материала и недостаток кислорода. Поэтому сгорание в нём идёт не полностью, и пламя этой зоны является “восстановительным”. Наконец, во внешнем конусе осуществляется полное сгорание при избытке кислорода воздуха, вследствие чего пламя здесь “окислительное”.
Так как добыча каменного угля весьма трудоёмка, Д. И. Менделеевым (1888 г.) была выдвинута идея подземной газификации угля. Сущность её заключается в получении газообразного горючего за счёт неполного сжигания угля под землей. В настоящее время подземная газификация угля является технологически освоенным процессом, но не находит широкого применения. Одной из причин этого является сравнительно малая калорийность получаемого газа (не выше 4200 кДж/м3). В будущем подземная газификация может приобрести большое значение для использования маломощных пластов, составляющих почти три четверти всех известных на Земле угольных запасов.
Интересно, что очень тонкая каменноугольная пыль вызывает нарушение устойчивости облаков не хуже сухого льда. Дешевизна этого материала позволит, вероятно, сильно расширить использование искусственного дождевания.
Постепенное обогащение атмосферы кислородом создало предпосылки для развития на поверхности Земли животной жизни. Около 350 млн. лет тому назад из животных форм океана развиваются первые предки современных нам земноводных, а около 250 млн. лет тому назад — пресмыкающиеся. В эпоху наибольшего господства последних, приблизительно 150 млн. лет тому назад, появляются первые предки современных птиц и несколько позднее — млекопитающих. Дальнейшая эволюция животных форм земной поверхности идёт в сторону постепенного вымирания земноводных и пресмыкающихся с заменой их более высокоорганизованными птицами и млекопитающими. В числе последних около 10 млн. лет тому назад развивается отдалённый предок современного человека.
Основная химическая реакция, доставляющая животным организмам необходимую им для жизни энергию, осуществляется в процессе дыхания и протекает по простой суммарной схеме:
С + О2 = СО2 + 393 кДж.
В результате этой реакции при жизнедеятельности организмов из атмосферы постепенно извлекается кислород и ей возвращается углекислый газ, чем и создаётся некоторый противовес процессу поглощения СО2 и выделения кислорода при росте растений. Экзотермическая реакция окисления углерода до СО2 протекает в тканях живого организма, куда углерод доставляется в виде органических веществ, извлекаемых из пищи. Необходимый для дыхания кислород поступает в организм человека через лёгкие, тонкие (0,004 мм) влажные стенки которых с громадной общей поверхностью (порядка 90 м2 при вдохе и 30 м2 при выдохе) позволяют этому газу проникать в систему обволакивающих лёгкие кровеносных сосудов. Здесь кислород образует непрочное химическое соединение с заключающимся в красных кровяных шариках сложным органическим веществом — гемоглобином — и в таком виде током красной артериальной крови разносится по тканям тела. В последних кислород отщепляется от гемоглобина и окисляет органические вещества пищи, причём получающийся углекислый газ частично образует нестойкое соединение с гемоглобином, главным же образом просто растворяется в кровяной жидкости и затем током тёмной венозной крови приносится в лёгкие, где СО2 и выделяется из организма.
В целом процесс дыхания может быть схематически изображён следующим образом (Гем — гемоглобин):
Гем + О2 = Гем·О2 (лёгкие: вдыхание)
Гем·О2 + С(из пищи) = Гем·СО2 (ткани)
Гем·СО2 = Гем + СО2 (лёгкие: выдыхание).
Таким образом, гемоглобин ведёт себя в процессе дыхания как катализатор. Частица его при молекулярном весе 68000 содержит 4 атома Fe, каждый из которых способен связывать одну молекулу О2.
Вдыхаемый воздух содержит приблизительно 21 объёмн. % О2, выдыхаемый — 16% кислорода и 4% СО2. В состоянии покоя человек потребляет около 20 л кислорода за час, и дыхание обеспечивает насыщение им артериальной крови до 95%. При снижении этого процента по тем или иным причинам (уменьшение парциального давления кислорода, дефекты самого дыхательного аппарата и др.) появляются симптомы кислородного голодания: понижение внимания, мышечная слабость, одышка и др.
За сутки через органы дыхания человека проходит около 20 м3 воздуха и он выдыхает 0,5 м3 углекислого газа. Для того, чтобы содержание этого газа в воздухе жилых помещений не поднималось выше 0,1%, необходимо их вентилировать, вводя за час около 20 м3 свежего воздуха на человека и уводя соответствующее количество “испорченного”. Обычно это осуществляется естественным путем сквозь щели, поры стен и за счет “проветривания”. В общественных помещениях, заводских цехах и т. д. применяется искусственная вентиляция.
Искусственная вентиляция становится особенно необходимой тогда, когда в воздухе заводских цехов может происходить накопление вредных для человека паров и газов, в частности СО. Этот газ реагирует с гемоглобином крови аналогично кислороду, причём образующееся соединение (Гем·СО) значительно более устойчиво. Поэтому даже при небольших концентрациях СО в воздухе значительная часть гемоглобина оказывается связанной с ним и, следовательно, перестаёт участвовать в переносе кислорода. Опыт показывает, что уже при содержании в воздухе 0,1 объёмн. % СО, т.е. при соотношении СО и кислорода 1:200, гемоглобином связываются равные количества обоих газов. Таким образом при вдыхании отравленного СО воздуха смерть от удушья может наступить, несмотря на наличие избытка кислорода. Ввиду обратимости реакции связывания гемоглобином как кислорода, так и СО, вдыхание “угоревшим” свежего воздуха (ещё лучше — чистого кислорода) ведёт к обратному выделению СО через лёгкие и постепенной замене его кислородом, что внешне проявляется в выздоровлении пострадавшего.
Интересные результаты были получены при потреблении напитков, насыщенных кислородом. Оказалось, что такое дополнительное его введение тонизирует весь организм, снимает чувство усталости и даёт положительный эффект при различных заболеваниях.
Подобно растительной, животная жизнь минувших эпох также оставила нам ценное наследство — нефть. Химизм образования нефтей ещё не вполне выяснен, всё же почти несомненно, что основным материалом для большинства из них послужили останки жизни мелководных морских бассейнов. Бурное развитие растительности (главным образом простейших водорослей), аналогичное “цветению” современных озёр, вело к столь бурному развитию животной жизни. Колоссальная быстрота размножения простейших организмов при благоприятных условиях привела к скоплению во впадинах дна водоёмов минувших эпох сотни тысяч тонн их останков. Медленно разлагаясь без доступа воздуха в стоячей придонной воде, останки эти постепенно заносились глиной и песком. На протяжении миллионов лет они превращались в нефть, причём углерод их выводился из круговорота.
Для характеристики поразительной скорости размножения простейших организмов можно привести пример: зелёная диатомовая водоросль при наиболее благоприятных условиях способна за месяц дать 2·1019 тонн вещества, т.е. массу, равную массе всего поверхностного слоя Земли в 16 км толщиной. Хотя в действительности скорость размножения простейших организмов строго ограничивается растительными условиями среды (содержание растворённых газов, элементов пищи и т. д.), она всё же очень велика.
Современные океаны и моря содержат громадные скопления подобных простейших организмов в верхних слоях воды до глубины примерно 200 м (планктон) и в придонной области не очень глубоких мест (бентос). Общее наличное количество планктона оценивается в 36 млрд. т. живого вещества, а бентоса — в 8 млрд. т. Будучи в конечном счёте основой питания всех сложных морских организмов, планктон и бентос вряд ли накапливаются теперь в форме своих останков. Иначе складывалось положение в минувшие эпохи, когда условия для развития простейших организмов были более благоприятными, а потребителей планктона и бентоса существовало значительно меньше.
Не исключена возможность и того, что в отдельных случаях исходным материалом для образования нефти послужили останки более высокоорганизованных животных (рыб и др.), массами гибнувших вследствие тех или иных причин. Экспериментально было показано, что при нагревании животного жира без доступа воздуха до высоких температур и под большим давлением из него образуются продукты, похожие по свойствам на обычные нефти. Есть мнение, что основным материалом для образования нефтей послужили не животные, а растительные организмы мелководных частей древнего моря.
Кроме рассмотренной “органической” теории происхождения нефти, являющейся ныне почти общепринятой, были предложены ещё две: “космическая” и “минеральная”. Согласно первой, нефти образовались в результате сжижения углеводородов, имевшихся ещё в первичной земной атмосфере. Теория эта представляется весьма маловероятной. Согласно “минеральной” теории, выдвинутой Д. И. Менделеевым (1876 г.), нефти образуются в результате взаимодействия проникающей в недра земли воды с раскаленными карбидами металлов. Теория эта сама по себе не представляется невероятной, однако тщательное изучение состава и свойств нефтей говорит против неё.
Обычные условия залегания нефтей говорят в пользу “органической” теории. Месторождения нефти встречаются в осадочных породах различного возраста. Скопления нефти располагаются под куполами пласта глины или другой водонепроницаемой породы. Над нефтью обычно находится скопление “нефтяного” газа, под ней — насыщенный соленой водой пласт песка.
Сырая нефть представляет собой нерастворимую в воде маслянистую коричневую или чёрную жидкость с зеленоватым отливом и плотностью 0,75-0,95 г/см3. По элементарному химическому составу она содержит 83-87% углерода, 14-11% водорода и небольшие количества азота, кислорода, серы (иногда также фосфора). Как показывают уже приведённые данные элементарного анализа, нефти состоят в основном из смеси различных углеводородов. В одних сортах преобладают члены гомологического ряда метана, в других — циклические углеводороды.
Нефть является очень ценным химическим сырьём, а также прекрасным топливом (1 кг даёт при сжигании около 46000 кДж). На нефтеперегонных заводах из неё выделяют ряд продуктов: петролейный эфир, бензин, лигроин, керосин, различные масла, вазелин, парафин и некоторые другие. Все эти вещества представляют собой смесь различных углеводородов от легколетучих (в петролейном эфире) до твёрдых при обычных условиях (в парафине). Очищенный керосин является одним из основных видов горючего жидких реактивных топлив. Нефтяной газ состоит в основном из газообразных углеводородов и может быть использован как в качестве топлива, так и для каталитического получения из него различных продуктов (водорода, спирта, формальдегида и др.). Вода нефтяных месторождений часто содержит значительные количества иода и брома и служит исходным сырьём для их добычи.
Широкое распространение двигателей внутреннего сгорания вызвало громадный рост потребления продуктов переработки нефти. За столетие с 1860 по 1960 г. её ежегодная мировая добыча возросла от 67 тыс. т. до 1 млрд. т. и продолжает быстро увеличиваться.
При оценке качества моторного топлива большое значение имеет его октановое число, определяющее режим работы мотора на данном топливе.
Работа двигателя внутреннего сгорания основана на использовании энергии периодических взрывов смеси паров горючего вещества с воздухом. Взрывы эти осуществляются в цилиндрах двигателя, где газовая смесь, после предварительного сжатия порциями, поджигается при помощи электрических искр. Чем сильнее сжата смесь перед взрывом, тем больше развиваемая мотором мощность. Однако практически сжатие можно осуществить только до известного предела, так как в дальнейшем происходит детонация газовой смеси, т.е. её взрыв с чрезмерно большой скоростью разложения. Допустимая степень сжатия при данном топливе и характеризуется его октановым числом. Чем оно больше, тем сильнее может быть сжата газовая смесь перед её взрывом и тем выше качество данного моторного топлива.
При построении условной шкалы октановых чисел значение 100 приписывают изооктану (СН3)3ССН2СН(СН3)2 (смесь паров которого с воздухом детонирует лишь при высокой степени сжатия), и значение 0 — легко детонирующему в парах нормальному гептану. Смешивая оба углеводорода в определённых соотношениях, получают отвечающие промежуточным точкам шкалы жидкости, с которыми экспериментально и сравнивают испытуемое топливо.
Величина октанового числа жидкого топлива сильно зависит от состава и строения входящих в него соединений. У обычных бензинов она редко превышает 70. Для повышения допустимых степеней сжатия к бензину часто добавляют небольшие количества (до 0,3%) антидетонаторов, наиболее известным из которых является тетраэтилсвинец — Pb(C2H5)4.
Процессы образования ископаемых каменных углей (особенно торфа) и нефти несомненно идут на отдельных участках земного шара и теперь, хотя, конечно, уже далеко не в столь больших масштабах, как раньше. Они продолжают играть некоторую роль и в современном нам круговороте углерода.
Из углекислого газа атмосферы и океана растениями извлекается ежегодно около 170 млрд. т. углерода. Значительная часть прироста растительной массы потребляется в пищу травоядными животными. Организмы последних служат, в свою очередь, пищей для плотоядных. Человек потребляет в пищу как животные, так и растительные продукты.
Было подсчитано, что в среднем каждые два месяца человек потребляет количество пищи, равное массе его тела. Расходуется она по двум направлениям:
1) на построение или обновление тканей и регулирование обмена веществ,
2) на производимую организмом работу и поддержание теплоты тела.
Для первого направления основное значение имеют белки и различные вещества, характеризующиеся небольшим содержанием их в пище (витамины, минеральные соли и т. п.). Функцию топлива в организме выполняют главным образом жиры и углеводы.
К оценке пищевых качеств какого-либо продукта приходится подходить, считаясь с обоими указанными выше факторами его значимости для организма. Кроме того, необходимо учитывать, что ни один пищевой продукт не усваивается полностью. В общем, пищевые вещества животного происхождения усваиваются человеком лучше, чем растительные. При приблизительной оценке доставленной организму теплоты можно в среднем считать, что каждый грамм пищевого белка даёт 19 кДж, жира — 38 кДж и углевода — 17 кДж. Питательная ценность некоторых пищевых продуктов с точки зрения развиваемого при их сжигании в организме тепла:
Продукт Состав, вес. % кДж на
вода белки жиры углеводы неусваем. ост. 100 г.
Белый хлеб 40,8 6,9 0,7 47,8 3,8 960
Хлеб ржаной 48,3 4,7 0,7 39,2 7,1 780
Говядина 70,4 19,0 9,5 0,0 1,1 695
Рыба (щука) 79,5 17,9 0,6 0,0 2,0 330
Молоко 87,6 3,3 3,5 4,4 1,2 270
Масло (сливочное) 15,5 0,5 79,3 0,5 4,2 3100
Сало (свиное) 0,7 0,2 95,1 0,0 4,0 3700
Сыр 45,5 22,6 20,0 3,4 8,5 1220
Количество энергии, которое должно быть получено человеческим организмом за счёт, пищи сильно зависит от климата, рода занятий, массы тела, пола, возраста и т. д. В очень грубо взятом среднем оно составляет 12500 кДж за сутки. С точки зрения лучшей переработки организмом средний суточный рацион целесообразно распределять приблизительно следующим образом: 100 г белков, 100 г жиров, 400 г углеводов. Жиры и углеводы могут быть без ущерба частично заменены друг другом. Напротив, белки в значительной части заменить жирами или углеводами нельзя, так как их основная роль существенно иная.
От характера потребляемой пищи до некоторой степени зависит рН крови. Так, питание преимущественно фруктами и овощами несколько смещает его в щелочную сторону, а преимущественно белковое питание — в кислую.
Для правильной работы организма важно введение в него достаточного количества минеральных солей и витаминов. Первые входят в состав почти всех видов пищевых продуктов и частично вводятся дополнительно (соление пищи). Витамины представляют собой сложные органические вещества, содержание которых в отдельных видах пищи очень различно. При недостаточном введении в организм витаминов нарушается обмен веществ и развиваются те или иные заболевания.
Высокую питательную ценность имеет молоко. По общей калорийности и пищевому составу литр молока заменяет 6 яиц. Молоко является почти единственным продуктом, содержащим одновременно все необходимые для организма витамины и минеральные соли. Особенно возрастает ценность молока при растительной диете.
Для обеспечения хорошего усвоения пищи необходимо разнообразить её, а также приправлять различными вкусовыми и пахучими веществами, вызывающими усиленное выделение пищеварительных соков. Существенно важно, что каждый орган человека имеет свой характерный режим питания. Так, мозг для нормальной работы нуждается преимущественно в сахаре, селезёнка — в гликогене (животном крахмале) и т. д. В общем можно сказать, что пища только тогда даёт максимальный полезный эффект, если она разнообразна по составу и вкусно приготовлена. Вопросом о качестве пищи не следует пренебрегать: “высокомерное невнимание к еде есть неблагоразумие” — И. П. Павлов.
Исключительную пищевую ценность могут иметь некоторые одноклеточные водоросли (хлорелла и др.). Так, в условиях достаточного азотного питания хлорелла содержит 50% белка (с хорошим аминокислотным составом), 35% углеводов (из которых только несколько процентов приходится на клетчатку), 5% жира, около 10% минеральных солей и все необходимые организму витамины. Опыты массового воспроизводства таких водорослей дали обнадёживающие результаты.
Замечательно то, что изменением условий питания, температуры и освещения можно сильно варьировать органический состав хлореллы. Например, из одной и той же исходной культуры были получены водоросли, содержащие 58% белка, 37,5 — углеводов и 4,5 — жира или 8,7 — белка, 5,7 — углеводов и 85,6 — жира. Её можно использовать для получения искусственной и синтетической пищи.
Дыхание животных и растений и тление их останков постоянно возвращает атмосфере (и водам океана) громадные массы углерода в виде углекислого газа. Если бы не происходило побочных процессов, общее возвращаемое подобным образом количество СО2 должно было бы приблизительно равняться усвоенному за то же время растениями. Однако в действительности всегда имеет место некоторый вывод углерода за счёт частичной минерализации останков растений и животных с образований торфа, ископаемых углей, нефти и т. д. Поэтому круговорот углерода не является вполне обратимым процессом, и уже в его органической части намечается основная линия свободного развития истории этого элемента — постепенный переход его из атмосферы в минералы земной поверхности.
В том же направлении, но ещё гораздо более мощно действуют неорганические реакции, протекающие между углекислым газом атмосферы и различными горными породами. При выветривании последних некоторые содержащиеся в них металлы под действием СО2 переходят в средние и кислые соли, вымываемые затем водой, переносимые реками в океан и частично осаждающиеся в нём. Общее количество углекислого газа, связываемого ежегодно при выветривании горных пород, по ориентировочным подсчётам отвечает 2 млрд. т углерода.
Этот громадный расход СО2 не могут компенсировать различные свободно протекающие природные процессы, ведущие к обратному переводу углерода из минералов в атмосферу (извержения вулканов, газовые источники, действие образующейся при грозах НNO3 на известняки и т. д.). Таким образом, и в своей неорганической части круговорот углерода направлен к уменьшению содержания СО2 в атмосфере.
Продолжавшийся на протяжении многих миллионов лет постепенный вывод углерода из атмосферы привёл к тому, что теперь она содержит у земной поверхности в среднем только 0,03% СО2. Так как углекислый газ (и водяной пар) свободно пропускает на Землю тепловое излучение Солнца и сильно задерживает обратное излучение Земли, уменьшение содержания СО2 в атмосфере явилось одной из причин изменения климата земной поверхности. Было вычислено, что при полном исчезновении СО2 из атмосферы средняя температура земной поверхности понизилась бы по сравнению с современной на 21 град. Напротив, при удвоении содержания СО2 она повысилась бы на 4 град (что привело бы к усиленному таянию льдов и резкому повышению уровня мирового океана). Так как в минувшие геологические эпохи атмосфера содержала больше углекислого газа (и водяных паров), средняя годовая температура на Земле была выше, чем в настоящее время (+14 °С).
Различное отношение содержащихся в атмосфере молекул СО2 к тепловому излучению Солнца и Земли обусловлено различием самого излучения. В среднем на уровне моря до поверхности Земли доходит около 75% того количества солнечной энергии [4850 кДж/(м2·ч)], которое получалось бы при отсутствии атмосферы. Из достигающего земной поверхности излучения лишь значительно меньшая часть отражается (море отражает примерно 10%, поверхность суши — от 3 до 25% и только снег отражает 50-90% падающего света), а большая часть поглощается. Тогда как главная доля энергии, доставляемой земной поверхности Солнцем, приходится на лучи с длинами волн 400-1800 нм, обратное излучение Земли характеризуется длинами волн от 400 нм и выше, причём особое значение имеют длины волн около 15000 нм: они соответствуют области избирательного поглощения углекислого газа. Около 20% теплового излучения Земли приходится на “окно” в области 900-1300 нм и почти полностью теряется. В общем, Земля теряет излучением лишь около трети того количества тепла, которое она теряла бы при отсутствии защитного действия СО2 и Н2О.
Подобно углекислому газу и водяным парам атмосферы ведёт себя обычное стекло. При этом оно не только само поглощает тепловое излучение Земли, но и изолирует прилегающей к ней слой атмосферы. Тем самым создаётся возможность без применения искусственного отопления поддерживать в оранжереях и парниках температуру значительно более высокую, чем в окружающем воздухе. Ещё лучшие результаты в том же направлении дают плёнки из ацетилцеллюлозы, полиэтилена и некоторых других пластмасс. Изыскание веществ и материалов, характеризующихся резко различным отношением к поглощению солнечного и земного излучения, составляет одну из важных задач, так как позволяет максимально использовать солнечную энергию и рационально разрешить ряд проблем народного хозяйства (перераспределение культурных растений в климатических поясах, лучшее прогревание жилищ в холодных областях и охлаждение в жарких и т. д.).
Развитие сознательной деятельности человека оказало влияние на все направления процессов, протекающих при свободном круговороте углерода. Вырубка лесных массивов, частичная замена их полями культурных растений и ряд подобных же изменений, внесённых в природу, не мог не сказаться на масштабах усвоения СО2 воздуха растениями и растительных организмов животными. Промышленное использование растительных и животных останков, а также потребление их в виде топлива (дрова, отчасти жиры и масла) в общем ускорило возвращение СО2 атмосфере. Косвенно деятельность человечества затронула и процессы минерализации растительных и животных останков, несколько ослабив их. Промышленная выработка полезных ископаемых, при которой образуется много минеральной пыли и обнажаются свежие слои горных пород, создаёт более благоприятные условия для их выветривания.
Все перечисленные линии сознательного воздействия человека отчасти компенсируют друг друга и не сказываются заметно на общем балансе круговорота углерода. Напротив, чрезвычайно сильно влияет на него увеличение потребления ископаемого минерального топлива. За счёт сжигания только одного каменного угля атмосфере ежегодно возвращается в виде СО2 более 2 млрд. т. углерода. Принимая во внимание потребление и других видов ископаемого горючего (нефти, газа, торфа и т. д.), а также ряд промышленных процессов, ведущих к выделению СО2 (например, обжиг известняка), можно думать, что человечество в настоящее время ежегодно вводит в круговорот около 3 млрд. т. углерода, заключённого до этого в минералах.
Таким образом, влияние человека на цикл превращений углерода по своему направлению прямо противоположно суммарным результатам его свободного развития.
Наиболее мощно действующим природным процессом, выводящим углерод из круговорота, является связывание СО2 при разрушении горных пород. Он ежегодно извлекает из атмосферы около 2 млрд. т. углерода. Но ещё больше этого элемента возвращает ей сознательная деятельность человека.
Общее количество углерода земной коры (трёх оболочек) составляет около 1017 т. причём большая его часть рассеяна повсюду в природе и поэтому не может быть даже ориентировочно распределена по отдельным формам нахождения:
Атмосфера 1·1012 т. Каменные угли 2·1013 т.
Океан 1·1014 т. Известняки 3·1016 т.
Живое вещество 1·1012 т.
Уже из её далеко не полных цифр видно, какие громадные массы этого элемента были на протяжении его земной истории выведены из круговорота в результате отложения каменных углей и известняков. Действительное количество углерода, извлеченное из первичной атмосферы, должно быть ещё значительнее, так как и большая часть его рассеянных соединений образовалось несомненно за счёт углекислого газа. Таким образом, в настоящее время атмосфера содержит лишь ничтожную часть того запаса СО2, который первоначально содержался в ней. Вместе с тем сопоставление данных ряда анализов воздуха, выполненных в разных местах и в разное время, приводит к выводу, что содержание СО2 в современной нам атмосфере медленно, но постоянно возрастает.
21
ФОСФОР
Весьма распространённый элемент; на его долю приходится около 0,04% от общего числа атомов земной коры. Он входит в состав некоторых белковых веществ (в частности, нервной и мозговой тканей), а также костей и зубов. Скопления фосфора встречаются главным образом в виде минерала апатита (Са5Х(РО4)3, где Х — F, реже Cl или ОН) и залежей фосфоритов, состоящих из фосфата кальция с различными примесями.
Фосфор открыт в 1669 г. Он является “чистым“ элементом — состоит только из атомов Р31.
В основном состоянии атом фосфора имеет структуру внешнего электронного слоя 3s23p3 и трёхвалентен. Сродство атома фосфора к электрону оценивается в 84 кДж/моль.
Минеральной основой костей является гидроксоапатит Са5(ОН)(РО4)3, а зубов — более твёрдый фторапатит Са5F(PO3)3. Общее содержание фосфора в человеческом организме составляет около 1 вес. %.
Значительные количества соединений фосфора (наряду с азотными) содержатся в экскрементах животных. Так, каждая тонна навоза содержит около 3 кг солей фосфорной кислоты (а с мочой человека их выделяется ежедневно около 4 г). Ещё больше фосфора содержат экскременты питающихся рыбой морских птиц. В результате жизнедеятельности громадных их стай на некоторых островах океана образуются залежи птичьих экскрементов (“гуано”), являющиеся объектом промышленного использования в качестве прекрасного удобрения.
Под действием дождей азотные соединения из гуано вымываются, а бульшая часть производных фосфора остаётся на месте, постепенно образуя залежи фосфоритов. Поэтому фосфориты могут образовываться и в местах массовой гибели различных животных. Такое происхождение (из экскрементов или трупов животных) для отдельных месторождений фосфоритов доказано. Другие месторождения образовывались в результате жизнедеятельности “фосфоробактерий”, массовое развитие которых имело место в некоторых древних морях. Наконец, существуют месторождения фосфоритов, для которых вероятно чисто минеральное происхождение.
Фосфориты и апатиты являются исходными продуктами для получения фосфорных минеральных удобрений, поэтому достаточные запасы этих минералов чрезвычайно важны для развития сельского хозяйства страны. В России известен ряд крупных месторождений фосфоритов, а на Кольском полуострове имеются громадные месторождения апатита.
Апатит представляет собой минерал неорганического происхождения и находит многообразное использование.
Свободный фосфор получают из природного фосфата кальция, прокаливая его с песком (SiO2) и углём в электрической печи. Процесс протекает по суммарной схеме:
Са3(РО4)2 + 3 SiO2 + 5 C + 1409 кДж = 3 СаSiO3 + 5 CO + 2 P.
Пары фосфора отводятся в орошаемые водой конденсаторы и затем собирают в приёмнике с водой, под слоем которой расплавленный фосфор и накапливается.
Процесс получения элементарного фосфора протекает около 1500 °С и идёт через две основные стадии. Сначала по уравнению:
Са3(РО4)2 + 8 С = 8 СО + Са3Р2
образуется фосфид кальция, который реагирует затем с Са3(РО4)2, давая СаО и фосфор:
3 Са3(РО4)2 + 5 Са3Р2 = 24 СаО + 16 Р.
Как показывает опыт, присутствие в исходной смеси SiO2, ннобязательно для протекания процесса, вместе с тем значительно его ускоряет за счёт понижения температуры плавления Са3(РО4)2 и связывания СаО по реакции:
СаО + SiO2 = CaSiO3.
Необходимое для получения фосфора тепло может быть сообщено системе не только за счёт электроэнергии (около 15 тыс. кВт·ч на тонну Р), но и за счёт сжигания кокса. При гораздо реже применяемом втором варианте процесс проводят в печах типа доменных. Ежегодная мировая выработка фосфора превышает 500 тыс. т.
В парах фосфор четырёхатомен, причём молекула Р4 имеет структуру правильного тетраэдра. Для твёрдого фосфора известно несколько аллотропных модификаций, из которых на практике приходится встречаться с двумя: белой и красной.
При охлаждении паров фосфора получается белая форма. Она образована молекулами Р4 и характеризуется плотностью 1,8 г/см3, температурой плавления 44 °С и температурой кипения 257 °С. В воде белый фосфор нерастворим, но хорошо растворим в сероуглероде (СS2). Хранят его под водой и по возможности в темноте.
При хранении белый фосфор постепенно (очень медленно) переходит в более устойчивую красную модификацию. Переход сопровождается выделением тепла:
Рбелый = Ркрасный + 17 кДж.
Процесс ускоряется при нагревании, под действием света и в присутствии следов иода.
Практически красный фосфор получают длительным нагреванием белого до 280-340 °С (в замкнутом объёме). Он представляет собой порошок с плотностью 2,3 г/см3, нерастворимый в сероуглерода и при нагревании возгоняющийся (т. возг. 429 °С). Пары его, сгущаясь, дают белый фосфор. Последний, в противоположность красному, очень ядовит.
В жидком или растворённом состоянии (как и в парах при температуре ниже 1000 °С) фосфор четырёхатомен. Энергия связи Р-Р в молекуле Р4 составляет 200 кДж/моль. Выше 1000 °С становится заметной диссоциация по схеме:
Р4 + 230 кДж = 2 Р2.
Содержащийся в молекулах РєР тройная связь характеризуется длиной 190 пм и энергией 489 кДж/моль. Дальнейший распад молекулы Р2 на атомы наступает лишь выше 2000 °С. Теплота атомизации фосфора (при 25 °С) равна 318 кДж/моль. Ниже -78 °С обычный белый фосфор превращается в другую, также бесцветную модификацию с плотностью 1,9 г/см3. При нагревании белой формы под давлением 500 атм образуется фиолетовый фосфор с плотностью 2,34 г/см3. Он имеет полимерную структуру. Тройной точке на его диаграмме состояния соответствуют температура 590 °С и давление 43 атм. Обычный красный фосфор представляет собой содержащее незначительные примеси мелкозернистое видоизменение фиолетового. Он известен в нескольких различных формах. При его возгонке в пар переходят молекулы Р2 (из которых затем образуются молекулы Р4). Теплота возгонки красного фосфора составляет 121 кДж/моль.
Выдерживание белого фосфора при 220 °С под давлением 12 тыс. атм (или под давлением 35 тыс. атм при 25 °С) может быть получен чёрный фосфор плотностью 2,7 г/см3. Теплота перехода в него белого фосфора составляет 38 кДж/моль. В присутствии ртути переход этот медленно осуществляется при 370 °С и без наложения высоких давлений (но полностью освободить конечный продукт от ртути не удаётся). Он имеет слоистую структуру. Он похож по внешнему виду на графит, обладает полупроводниковой проводимостью, а по химическим свойствам подобен красному фосфору(но на воздухе вполне устойчив и воспламеняется лишь выше 400 °С). Под давлением 18 тыс. атм чёрный фосфор плавится около 1000 °С, а под давлением только своего пара выше 550 °С переходит в фиолетовый. Выше 111 тыс. атм возникает металлическая фаза фосфора с простой кубической структурой.
Теплота плавления белого фосфора 2,5 кДж, а теплота его испарения 50 кДж (на моль Р4). Продаётся он обычно отлитым в палочки, которые легко режутся ножом. Эту операцию необходимо проводить под водой (лучше всего при 20-25 °С), так как при разрезании на воздухе фосфор может воспламениться от трения. По той же причине высушивать кусочки белого фосфора следует, прикладывая к ним полоски фильтровальной бумаги и избегая трения или надавливания. Ни в коем случае нельзя брать кусочки белого фосфора пальцами (а только щипцами или пинцетом).
Растворимость белого фосфора в сероуглероде исключительно велика (порядка 10:1 при обычных условиях). При медленном упаривании такого раствора фосфор выделяется в виде прекрасно образованных бесцветных кристаллов, Белый фосфор растворим и в ряде других органических жидкостей (бензоле, эфире и т. д.), а также в жидких SO2 и NH3. Технический продукт может быть очищен перекристаллизацией или перегонкой в атмосфере азота. Расплавленный белый фосфор весьма склонен к переохлаждению (капли диаметром в 1 мм удалось переохладить до -71 °С).
Средством первой помощи при отравлении фосфором служит 1%-ный раствор СuSO4 (по чайной ложке через каждые 5 мин до появления рвоты). Горящий фосфор причиняет болезненные и трудно заживающие ожоги, которые могут вызвать также общее отравление организма. Средством первой помощи при ожоге фосфором служит мокрая повязка, пропитанная 5%-ным раствором CuSO4.
Раствор CuSO4 рекомендуется и при тушении горящего фосфора. Действие его основано на восстановлении меди до металла (по схеме
2 Р + 5 CuSO4 + 8 H2O = 2 H3PO4 + 5 Cu + 5 H2SO4),
плёнка которого обволакивает ещё не окислившийся фосфор. По сути дела, реакция эта аналогична вытеснению меди цинком наглядно демонстрирует наличие у элементарного (белого) фосфора электродной функции.
Хотя красный фосфор окисляется несравненно труднее белого, однако его медленное взаимодействие с кислородом воздуха всё же происходит (особенно — в присутствии следов Fe или Cu). Результатом этого является образование незначительных количеств очень гигроскопичных продуктов окисления и “отмокание” красного фосфора при его хранении в неплотно закупоренных банках. Отмокший красный фосфор перед употреблением следует перенести на фильтр, тщательно промыть водой и сушить в сушильном шкафу. От примеси белого фосфора красный может быть очищен длительным кипячением с 7%-ным раствором едкого натра и затем с водой.
Наибольшие количества свободного (красного) фосфора потребляются спичечным производством. Соединения фосфора используются главным образом в виде минеральных удобрений.
Белый фосфор очень ядовит, поэтому употребление его для выработки спичек (воспламеняющихся при трении о любую твёрдую поверхность) запрещено. Обычные спички изготавливаются на основе красного фосфора. Они воспламеняютая только при трении о специально подготовленную поверхность (намазку спичечной коробки). Существуют различные рецепты их изготовления. Примером может служить приводимый ниже состав (в вес. %):
| Головка Намазка |
| Бертолетова соль 46,5 Красный фосфор 30,8 |
| Хромпик 1,5 Трёхсернистая сурьма 41,8 |
| Сера 4,2 Сурик или мумия 12,8 |
| Цинковые белила 3,8 Мел 2,6 |
| Сурик или мумия 15,3 Цинковые белила 1,5 |
| Молотое стекло 17,2 Молотое стекло 3,8 |
| Клей костяной 11,5 Клей костяной 6,7 |
При трении головки о намазку мельчайшие частички фосфора воспламеняются на воздухе и поджигают состав головки. Для уменьшения пожарной опасности осиновая древесина (“соломка”) спичек при их выработке примерно до половины пропитывается раствором фосфорнокислого аммония, вследствие чего они гаснут без последующего тления.
Химическая активность фосфора значительно выше, чем у азота. Так, он легко соединяется с кислородом, галогенами, серой и многими металлами. В последнем случае образуются аналогичные нитридам фосфиды (Mg3P2, Ca3P2 и др.).
Белый фосфор значительно более реакционноспособен, чем красный. Так, он медленно окисляется на воздухе даже при низких температурах и воспламеняется уже выше 50 °С, тогда как красный фосфор на воздухе почти не окисляется, а воспламеняется лишь при более высокой температуре. Точно так же и другие реакции протекают с белым фосфором энергичнее, чем с красным. Подобное различие реакционной способности аллотропных модификаций является общим случаем: из двух форм одного и того же вещества менее устойчивая обычно более активна.
Как и в случае азота наиболее характерными валентными состояниями фосфора -3, 0, +3 и 5.
С водородом фосфором практически не соединяется. Однако разложением некоторых фосфидов водой по реакции, например
Са3Р2 + 6 Н2О = 3 Са(ОН)2 + 2 РН3
может быть получен аналогичный аммиаку фосфористый водород (фосфин) — РН3. Последний представляет собой бесцветный газ с неприятным запахом (“гнилой рыбы”). Фосфин является очень сильным восстановителем и весьма ядовит. В противоположность аммиаку реакции присоединения для фосфина мало характерны; соли фосфония (РН4+) известны лишь для немногих сильных кислот и весьма нестойки, а с водой фосфин химически не взаимодействует (хотя довольно хорошо растворим в ней).
Реакция взаимодействия фосфора (белого) с водородом слабо экзотермична:
2 Р + 3 Н2Ы 2 РН3 + 12,5 кДж.
С заметной скоростью она протекает лишь выше 300 °С, когда выход фосфина не превышает долей процента. Применением высоких давлений он может быть несколько повышен, но всё же при 350 °С и 200 атм составляет только 2%. Равновесие в этих условиях устанавливается лишь через 6 суток.
Молекула РН3 полярна и имеет структуру треугольной пирамиды с атомов Р в вершине. Фосфин (т. пл. -133, т. кип. -88 °С) довольно неустойчив, но при обычных температурах самопроизвольно не разлагается. На воздухе он воспламеняется около 150 °С. При отравлении фосфином прежде всего страдает нервная система (одышка, слабость, конвульсии). В качестве средства первой помощи рекомендуется вдыхание кислорода. Предельно допустимой концентрацией РН3 в воздухе производственных помещений считается 1·10-4 мг/л.
Растворимость фосфина в воде составляет около 1:4 по объёму (в органических растворителях она значительно выше). Для него известен очень нестойкий кристаллогидрат РН3·Н2О, по составу отвечающий гидроксиду фосфония (РН4ОН). Электролитическая диссоциация фосфина ничтожно мала и имеет амфотерный характер: для реакций по схемам:
РН3 + Н2О Ы РН4· + НО’ и
РН3 + Н2О Ы РН2’+ Н3О•
были найдены значения констант равновесия соответственно 4·10-29 и 2·10-29.
Содержащие в своём составе тетраэдрические ионы РН4+, соли фосфония представляют собой бесцветные кристаллические вещества. Перхлорат фосфония (РН4СlO4) весьма взрывчат, а галогениды возгоняются, причём в парах они практически полностью диссоциированы на РН3 и соответствующий галогенводород. Термическая их устойчивость несравненно меньше, чем у аналогичных солей аммония, как это видно из приводимого сопоставления температур, при которых, при которых давление возникающих в результате диссоциации паров достигает одной атмосферы:
NH4Cl | NH4Br | NH4I | PH4Cl | PH4Br | PH4I | |
| 339 | 388 | 400 | -28 | 35 | 62 |
Подобно самому РН3, галогениды фосфония являются очень сильными восстановителями. Водой они разлагаются на фосфин и соответствующую галогенводородную кислоту. Особенно легко идёт подобный распад в присутствии щёлочи, чем пользуются при получении чистого РН3. Другим методом получения чистого фосфина может служить нагревание белого фосфора с крепким спиртовым раствором КОН.
Продукты частичного замещения водородов РН3 на металл плохо изучены. В частности NaPH2 может быть получен взаимодействием РН3 с раствором металлического натрия в жидком аммиаке и представляют собой белое твёрдое вещество с ионной структурой. На воздухе NaPH2 самовоспламеняется, при нагревании в вакууме до 100 °С он переходит в Na2PH по реакции:
2 NaPH2 = PH3 + Na2PH,
а водой тотчас разлагается на PH3 и NaOH. Интересна протекающая в водном растворе реакция по уравнению:
РН3 + 3 HgCl2 = P(HgCl)3Ї + 3 HCl,
которая может быть использована для количественного определения фосфина.
В качестве продуктов полного замещения водородов РН3 на металлы можно рассматривать их фосфиды, хотя состав последних, как и у нитридов далеко не всегда отвечает валентным соотношениям. Например, для уже рассмотренных металлов описаны соединения следующих составов: Э3Р (Mn, Re, Cr, Mo), Э2Р (Mn, Re, Cr, W), ЭР (Mn, Re, Cr, Mo, W), ЭР2 (Mn, Re). Подобно нитридам, многие весьма устойчивые по отношению не только к воде, но и к кислотам.
Наряду с РН3,при разложении водой фосфидов всегда образуется небольшое количество дифосфина — Р2Н4. Это бесцветная жидкость (т. пл. -99, т. кип. 63 °С). По строению молекула подобна гидрозину. При взаимодействии белого фосфора с щелочными металлами в жидком аммиаке образуются солеобразные продукты звмещения водорода, имеющие оранжевую окраску. С кислотами дифосфин (“жидкий фосфористый водород”) не реагирует, а на воздухе самовоспламеняется.
При хранении Р2Н4 постепенно распадается на РН3 и аморфное твёрдое вещество жёлтого цвета, которому приписывались формулы Р12Н6 или Р5Н2. Описан также оранжевый гидрид состава Р9Н2. Эти “твёрдые фосфористые водороды” представляют собой не определённые химические соединения, а растворы РН3 в белом фосфоре.
Вместе с тем может быть получен (например, взаимодействием LiH c эфирным раствором PСl3) жёлтый твёрдый полимер (РН)х. Он нерастворим во всех обычных растворителях, устойчив по отношению к щелочам и кислотам, а при нагревании выше 400 °С (в вакууме) разлагается по схеме:
6 РН = Р4 + 2 РН3.
Существует указание на возможность получения смеси высших фосфинов — цепеобразных РnНn+2 и циклических РnНn.
Одним из применяемых для получения РН3 методов является нагревание белого фосфора с крепким водным раствором щелочи. Реакция идёт, например, по уравнению:
8 Р + 3 Ва(ОН)2 + 6 Н2О = 2 РН3 + 3 Ва(Н2РО2)2.
Вторым продуктом этой реакции является бариевая соль фосфорноватистой кислоты.
Действием на эту соль серной кислотой может быть получена свободная фосфорноватистая кислота (Н3РО2). Несмотря на наличие в её молекуле трёх атомов водорода, она только одноосновна (и является довольно сильной), что согласуется со структурной формулой:
Н
Ѕ
Н—О—Р—Н
||
О
Соли фосфорноватистой кислоты (гипофосфиты) хорошо растворимы в воде.
Гипофосфит бария легко очищается перекристаллизацией. После его обменного разложения с Н2SO4 из сгущённого и охлаждённого фильтрата (от ВаSO4) фосфорноватистая кислота выделяется в виде больших кристаллов, плавящихся при 27 °С (и при дальнейшем нагревании разлагающихся). Она может быть получена взаимодействием РН3 с водной суспензией иода по схеме:
2 I2 + 2 Н2О + РН3 = 4 НI + H3PO2.
В растворе Н3PO2 проявляет тенденцию к распаду с выделением водорода и образованием Н3PO3 и H3PO4, но распад этот без катализаторов (Pd и т. п.) становится практически заметным лишь при высоких температурах или в сильнощелочной среде. При нагревании возможна также дисмутация по схеме:
2 Н3РО2 = Н3РО4 + РН3.
Водородом в момент выделения фосфорноватистая кислота (К = 9·10-2) восстанавливается до РН3. В сильнокислой среде (особенно при нагревании) она является очень энергичным восстановителем. Например, соли ртути восстанавливаются ею до металла:
HgCl2 + H3PO2 + H2O = H3PO3 + Hg + 2 HСl.
Напротив, в разбавленных растворах на холоду Н3РО2 не окисляется ни кислородом воздуха, ни свободным иодом.
Взаимодействие фосфора с кислородом в зависимости от условий ведёт к образованию различных продуктов. При сгорании фосфора в избытке кислорода (или воздуха) получается его высшей оксид — фосфорный ангидрид (Р2О5). Напротив, горение при недостатке воздуха или медленное окисление даёт главным образом фосфористый ангидрид (Р2О3).
Реакция медленного окисления фосфора кислородом воздуха интересна с различных сторон. Прежде всего, она сопровождается свечением, которое хорошо видно в темноте. Параллельно с окислением фосфора всегда происходит образование озона. Обусловлено это промежуточным возникновением радикала фосфорила (РО) по схеме: Р + О2 = РО + О и последующей побочной реакцией О + О2 = О3. Наконец, с окислением фосфора связана ионизация окружающего воздуха, что резко сказывается на его электропроводности. Этот эффект наблюдается и при некоторых других химических процессах, например при окислении на воздухе натрия или калия.
Выделение света при протекающих без заметного разогревания химических реакциях называется хемилюминесценцией. Она наблюдается не только при медленном окислении фосфора, но и при некоторых других химических и биохимических процессах, которыми обусловлено, в частности, свечение светляков, гнилушек и т. д. Зелёная хемилюминесценция фосфора во влажных средах связана с промежуточным образованием молекулы НРО.
Реакция окисления фосфора протекает только в известном интервале концентраций кислорода. При его парциальных давлениях ниже некоторого минимального (порядка 0,05 мм рт. ст.), а также выше некоторого максимального предела окисление практически не происходит. Сам интервал благоприятных для реакции концентраций зависит от температуры (и некоторых других факторов). Так, при обычных условиях скорость окисления фосфора чистым кислородом возрастает с увеличением его давления вплоть до 300 мм рт. ст., а затем начинает уменьшаться и при давлении 700 мм рт. ст. и выше становится близкой к нулю. Таким образом, в чистом кислороде фосфор при обычных условиях практически не окисляется. Само наличие нижней и верхней границ давления связано с цепным характером реакции окисления. Подобные случаи изучены также для мышьяка и серы.
Последний представляет собой белую, похожую на воск кристаллическую массу. При нагревании на воздухе он переходит в Р2О5 (а постепенно окисляется уже в обычных условиях). Взаимодействуя с холодной водой, Р2О3 медленно образует фосфористую кислоту:
Р2О3 + 3 Н2О = 2 Н3РО3.
Подобно белому фосфору, фосфористый ангидрид очень ядовит.
Фосфористый ангидрид (т. пл. 24, т. кип. 175°С) можно отделить от менее летучего Р2О5 отгонкой. В органических растворителях Р2О3 хорошо растворим.
Определения молекулярного веса дифосфортриоксида и в газообразном состоянии, и в растворах согласно приводят к удвоенной формуле. Теплота образования Р4О6 из элементов равна 1640 кДж/моль, а энергия связи Р-О оценивается в 360 кДж/моль. При взаимодействии Р4О6 с холодной водой образуется только Н3РО3, а с горячей водой и газообразным HСl реакции протекают в основном по уравнениям:
P4O6 + 6 H2O = PH3 + 3 H3PO4 и
P4O6 + 6 HCl = 2 H3PO3 + 3 PCl3.
Очень энергично реагирует фосфористый ангидрид также с хлором, бромом и серой (выше 150 °С).
Свободная фосфористаякислота (Н3РО3) представляет собой бесцветные кристаллы, расплывающиеся на воздухе и легкорастворимые в воде. Она является сильным (но в большинстве случаев медленно действующим) восстановителем. Несмотря на наличие в молекуле трёх водородов, Н3РО3 функционирует только как двухосновная кислота средней силы. Соли её (фосфористокислые или фосфиты), как правило, бесцветны и труднорастворимы в воде. Из производных чаще встречающихся металлов хорошо растворимы лишь соли Na, K и Са.
Строение фосфористой кислоты может быть выражено следующими структурными формулами:
![]()
![]()
![]() Н—О Н—О Н
Н—О Н—О Н
![]()
![]()
![]()
![]()
![]() Р—О—Н или Р
Р—О—Н или Р
Н—О Н—О О
![]() Фосфористую кислоту (т. пл. 74 °С, К1 = 6·10-2, К2
Фосфористую кислоту (т. пл. 74 °С, К1 = 6·10-2, К2
![]() Н = 2·10-7) удобно получать гидролизом
Н = 2·10-7) удобно получать гидролизом
![]() Н трёххлористого фосфора и последующим
Н трёххлористого фосфора и последующим
![]() упариванием жидкости до начала кристаллизации.
упариванием жидкости до начала кристаллизации.
![]()
![]()
![]()
![]() Р Установленное рентгеновским анализом кристалла
Р Установленное рентгеновским анализом кристалла
![]() О О строение молекулы Н3РО3 показано на рис. 1
О О строение молекулы Н3РО3 показано на рис. 1
(пунктирами отмечены водородные связи с
![]() О Н соседними молекулами).
О Н соседними молекулами).
Рис. 1 В растворе равновесие по схеме
НРО(ОН)2Ы Р(ОН)3
смещено влево, по-видимому, ещё гораздо значительнее, чем у фосфорноватистой кислоты. Вместе с тем было показано, что фосфористая кислота способна присоединять протон, причём для константы равновесия такой её функции даётся значение [H3PO3][H+]/[H4PO3+] = 105.
Водородом в момент выделения Н3РО3 восстанавливается до РН3, а при нагревании безводной кислоты или её концентрированных растворов происходит дисмутация по схеме:
4 Н3РО3 = РН3 + 3 Н3РО4.
Кислородом воздуха растворы фосфористой кислоты при обычных условиях заметно окисляются только в присутствии следов иода, а чистая (не содержащая оксидов азота) азотная кислота не окисляет её даже при кипячении. Взаимодействие фосфористой кислоты с хлоридом ртути (II) медленно идёт по уравнению:
Н3РО3 + 2 HgCl2 + H2O = H3PO4 + Hg2Cl2 + 2 HCl.
Для фосфористой кислоты известны не только средние, но и кислые соли. Примерами тех и других могут служить Na2HPO3·5H2O и NaH2PO3·2,5H2O. Получены также некоторые комплексные производные Н3РО3, например, зелёная кислота Н3[Cr(HPO3)3]·10H2O и некоторые её соли. При прокаливании фосфидов происходит распад их на соответствующие фосфаты и производные низших степеней окисления фосфора, вплоть до РН3.
Нагреванием NaH2PO3 при 150 °С в вакууме до прекращения выделения воды может быть получен Na2H2P2O5, представляющий собой соль пирофосфористой кислоты (H4P2O5). Свободная кислота (т. пл. 36 °С) была получена по схеме:
5 Н3РО3 + РCl3 = 3 HCl + 3 H4P2O5.
Она двухосновна и малоустойчива, имеет симметричное строение, выражаемое формулой НО(Н)(О)РОР(О)(Н)ОН. Растворы пирофосфита натрия в обычных условиях устойчивы, но при кипячении (или в кислой среде) происходит присоединение воды с образованием ортофосфита. Получены также соли, отвечающие метафосфористой кислоте (НРО2). В свободном состоянии она частично образуется при сгорании фосфина.
При окислении влажного фосфора кислородом наряду с Р2О3 и Р2О5 всегда образуется также фосфорноватая кислота — Н4Р2О6 (необходимость удвоения простейшей формулы доказано определением молекулярного веса и результатами изучения магнитных свойств), структура которой отвечает формуле (НО)2ОР-РО(ОН)2 с непосредственной связью между обоими атомами фосфора. От других кислот этого элемента фосфорноватую отделяют при помощи её труднорастворимой (2:100) соли Na2H2P2O6·6H2O. Последнюю удобно получать обработкой красного фосфора смесью Н2О2 и крепкого раствора NaOH. Ион Н2Р2О62- характеризуется параметрами d(PP) = 217, d(P-OH) = 157, d(P=O) = 150 пм.
Свободную кислоту выделяют обычно обменным разложением её почти нерастворимой бариевой соли с разбавленной серной кислотой. После упаривания раствора фосфорноватая кислота кристаллизуется в виде больших бесцветных пластинок состава Н4Р2О6·2Н2О, плавящихся при 62 °С. На воздухе эти кристаллы легко расплываются, тогда как собственную кристаллизационную воду они теряют лишь при длительном хранении в вакууме над Р2О5. Безводная Н4Р2О6 плавится при 73 °С (с разл.). Как кислота она средней силы (К1 > 10-2, К2 = 2·10-3, К3 = 5·10-8, К4 = 9·10-11). При хранении фосфорноватая кислота постепенно разлагается. В растворах на холоду она довольно устойчива, а нагревание сопровождается её распадом по схеме:
Н4Р2О6 + Н2О = Н3РО3 + Н3РО4,
причём процесс протекает тем быстрее, чем выше концентрация водородных ионов. Ангидрид фосфорноватой кислоты неизвестен. Оксид фосфора состава Р4О8 в качестве такового рассматривать нельзя, так как ни перехода от него к фосфорноватой кислоте, ни обратного перехода осуществить не удаётся.
Фосфорноватая кислота окисляется до фосфорной лишь при действии самых сильных окислителей (KMnO4 и т. п.). С другой стороны, сама она окислителем не является. Все четыре водорода фосфорноватой кислоты могут быть замещены на металл, причём образующиеся соли (гипофосфаты), как правило, бесцветны и труднорастворимы в воде. Хорошо растворяются лишь производные наиболее активных одновалентных металлов. Растворы их вполне устойчивы как у самой Н4Р2О6, как и у её солей сильно выражена склонность к реакциям присоединения.
Непосредственная связь между атомами фосфора имеется не только в Н4Р2О6, но и в молекулах некоторых других кислот фосфора. Сюда относятся Н4Р2О4, Н5Р3О9 и изомерная пирофосфористой трёхосновная Н4Р2О5 (по одной связи Р-Р), Н5Р3О8 и Н6Р4О11 (по две связи), циклические Н4Р4О10 (две связи) и Н6Р6О12 (шесть связей). Кислоты эти известны главным образом в виде своих солей.
Несколько лучше других изучена Н4Р2О4, строение которой отвечает, по-видимому, формуле НО(Н)ОР-РО(Н)ОН. Кислота эта была выделена в виде её малорастворимой бариевой соли ВаН2Р2О4 из продуктов гидролиза Р2I4. В растворах она легко окисляется.
В результате реакции по схеме:
РCl3 + H3PO4 = 3 HCl + H4P2O6
образуется фосфористофосфорная кислота, имеющая тот же общий состав, что и фосфорноватая. Как следует из её структурной формулы НО(Н)(О)Р-О-Р(О)(ОН)2, кислота эта трёхосновна. Она является главным продуктом разложения фосфорноватой кислоты при её хранении, а в растворе быстро гидролизуется. Фосфитофосфат натрия Na3HP2O6 может быть получен совместным нагреванием NaH2PO3 c Na2HPO4 при 180 °С.
Наиболее характерный для фосфора оксид — фосфорный ангидрид (Р2О5) представляет собой белый порошок. Он чрезвычайно энергично притягивает влагу и поэтому часто применяется в качестве осушителя газов. Вместе с тем Р2О5 во многих случаях отнимает от различных веществ также химически связанную воду, чем пользуются при получении некоторых соединений.
Теплота образования Р2О5 из элементов составляет 1490 кДж/моль. определение молекулярного веса фосфорного ангидрида в парах указывает на удвоенную формулу — Р4О10.
Твёрдый фосфорный ангидрид — (Р2О5)n — известен в трёх кристаллических модификациях. Первая, по виду похожа на снег, слагается из отдельных молекул Р4О10, связанных друг с другом лишь межмолекулярными силами, она довольно легко возгоняется (т. возг. 359 °С).
При нагревании этой формы до 400 °С в запаянной трубке получается полимерная форма, образованная бесконечными слоями тетраэдров РО4 с общими (тремя из четырёх) атомами кислорода.
Длительное выдерживание данной формы в запаянной трубке при 450 °С сопровождается её переходом в другую полимерную форму. Это наиболее устойчивая модификация фосфорного ангидрида.
Фосфорный ангидрид, поступающий в продажу, обычно представляет собой смесь первой и второй форм, более или менее загрязнённую примесями воды и продуктов неполного сгорания фосфора. Очистка Р2О5 осуществляется его возгонкой в быстром токе сухого кислорода (причём получается первая форма). Чистый фосфорный ангидрид совершенно не имеет запаха.
Ниже сопоставлены остаточные давления водяного пара (в мм рт. ст. при 20 °С) над некоторыми наиболее распространёнными осушителями. Чем меньше эти давления, тем энергичнее действует данный осушитель.
CuSO4 | ZnCl2 | CaCl2 | NaOH | H2SO4 | KOH | Mg(ClO4)2 | P2O5 |
| 1,4 | 0,8 | 0,36 | 0,16 | 0,003 | 0,002 | 0,0005 | 0,00002 |
Из сопоставления видно, что по интенсивности осушающего действия Р2О5 далеко превосходит все остальные вещества. Однако при пользовании техническим продуктом следует учитывать возможность загрязнения очищаемых газов фосфористым водородом (из-за наличия в Р2О5 примеси низших оксидов фосфора). Во многих случаях не исключена также возможность протекания при сушке химических процессов. Например, хлористый водород способен реагировать по схеме:
Р4О10 + 3 НСl = POCl3 + 3 HPO3.
Взаимодействие фосфорного ангидрида с водой идёт весьма энергично и сопровождается значительным выделением тепла (до 192 кДж/моль).
Термическое разложение Р4О6 сопровождается частичным отщеплением элементарного фосфора с образованием смесей оксидов состава Р4О7, Р4О8, Р4О9, по строению подобных Р4О10 (без части переферических атомов кислорода). Лучше изучены из них Р4О8 может быть индивидуально получен по схеме:
4 Р4О6 = Р4 + 3 Р4О8
длительным нагреванием тетрафосфоргексоксида в запаянной трубке несколько выше 210 °С. Он представляет собой блестящие бесцветные кристаллы, возгоняющиеся выше 180 °С, устойчивые по отношению к нагреванию (в отсутствие воздуха), нерастворимые в органических растворителях и медленно взаимодействующие с водой по схеме:
Р4О8 + 6 Н2О = 2 Р(ОН)3 + 2 Н3РО4.
Оксид этот является, следовательно, смешанным ангидридом фосфористой и фосфорной кислот.
Взаимодействие Р2О5 с водой в зависимости от числа присоединённых молекул Н2О приводит к образованию следующих основных гидратных форм:
Р2О5 + Н2О = 2 НРО3 (метафосфорная кислота)
Р2О5 + 2 Н2О = 2 Н4Р2О7 (пирофосфорная кислота)
Р2О5 + 3 Н2О = 2 Н3РО4 (ортофосфорная кислота).
Как видно из приведённого сопоставления, наиболее богата водой орто-кислота, которую обычно называют просто фосфорной. При её нагревании происходит отщепление воды, причём последовательно образуются пиро- и мета- формы:
2 Н3РО4 + 71 кДж = Н2О + Н4Р2О7 и
Н4Р2О7 + 100 кДж = Н2О + 2 НРО3.
При действии воды обе кислоты переходят в орто-форму. Однако переходы эти на холоду протекают крайне медленно. Поэтому каждая кислота реагирует в свежеприготовленном растворе как индивидуальное вещество.
Наибольшее практическое значение из кислот пятивалентного фосфора имеет ортогидрат (Н3РО4). Получать его удобно окислением фосфора азотной кислотой:
3 Р + 5 НNO3 + 2 H2O = 3 H3PO4 + 5 NO.
В промышленности Н3РО4 получают исходя из Р2О5, образующегося при сжигании фосфора (или его паров) на воздухе.
Интересен метод получения Н3РО4 путём взаимодействия паров фосфора и воды по реакции:
Р4 + 16 Н2О = 4 Н3РО4 + 10 Н2 + 1305 кДж,
в присутствии катализатора (например, мелкораздробленной меди) достаточно быстро протекающей около 700 °С. Как видно из уравнения, фосфор ведёт себя в данном случае подобно цинку или железу. Получающийся водород используется для синтеза NH3. Рассматриваемый процесс особенно пригоден для выработки аммофоса.
Фосфорная кислота представляет собой бесцветные, расплывающиеся на воздухе кристаллы. Продаётся она обычно в виде 85%-ного водного раствора, приблизительно отвечающего составу 2Н3РО4·Н2О и имеющего консистенцию густого сиропа. В отличие от многих других производных фосфора, фосфорная кислота неядовита. Окислительные свойства для неё вовсе не характерны.
Будучи трёхосновной кислотой средней силы, Н3РО4 способна образовывать три ряда солей, дигидрофосфаты, гидрофосфаты и фосфаты. Дигидрофосфаты хорошо растворимы в воде, а из гидрофосфатов и фосфатов растворимы лишь немногие, в частности соли Na. Как правило, фосфаты бесцветны.
Безводная фосфорная кислота (т. пл. 42 °С) весьма склонна к переохлаждению, а при нагревании заметно летуча. В жидком состоянии она характеризуется высоким значением диэлектрической проницаемости (e = 61 при 25 °С) и в ней довольно сильно представлена самодиссоциация по схеме:
2 Н3РО4Ы Н4РО4+ + Н2РО4-.
При нагревании она активно разъедает стекло и почти все металлы. Для неё известен кристаллогидрат 2Н3РО4·Н2О (т. пл. 30 °С). В водных растворах Н3РО4 умеренно диссоциирована (К1 = 7·10-3, К2 = 6·10-8, К3 = 4·10-13). Её 0,1 н раствор имеет рН = 1,5, максимальной электропроводностью обладает 48%-ный раствор, а 65%-ный раствор замерзает лишь около -85 °С. Фосфорная кислота используется иногда для изготовления прохладительных напитков.
Наличие у фосфорной кислоты заметных признаков амфотерности выявляется при её взаимодействии с НСlO4. Реакция (в отсутствие воды) идёт по уравнению:
РО(ОН)3 + НСlO4 = [P(OH)4]ClO4.
Получающееся солеобразное соединение представляет собой бесцветные кристаллы (т. пл. 47 °С). Подобным же образом взаимодействует Н3РО4 и с серной кислотой.
Разбавленные (1%-ные) растворы фосфатов натрия характеризуются следующими значениями концентраций водородных ионов: NaH2PO4 — pH = 4,6, Na2HPO4 — pH = 8,9 и Na3PO4 — pH = 12,1.
При прокаливании дигидрофосфаты образуют метафосфаты и выделяют воду. Гидрофосфаты при нагревании образуют пирофосфаты и воду, а фосфаты остаются без изменения. Если катион термически неустойчив, при прокаливании происходит распад соли с выделением летучих продуктов разложения. Например, из малорастворимого смешанного третичного фосфата магния и аммония MgNH4PO4 c выделением NH3 и Н2О образуется пирофосфорнокислый магний — Mg2P2O7. Образование малорастворимого MgNH4PO4 используется при количественном определении фосфорной кислоты (а также магния).
Пирофосфорная кислота образуется при постепенном нагревании ортофосфорной до 260 °С. Она представляет собой мягкую стекловидную массу (т. пл. 61 °С), легкорастворимую в воде. Обратный переход в ортогидрат идёт на холоду лишь очень медленно. При кипячении раствора, особенно в присутствии сильных кислот, он значительно ускоряется.
Пирофосфорная кислота четырёхосновна, причём по лёгкости диссоциации два первых её водорода резко отличаются от двух других (К1 = 3·10-2, К2 = 4·10-3, К3 = 3·10-7, К4 = 6·10-10). Ион Р2О74- построен из двух тетраэдров РО4 с одним общим кислородным атомом [d(PO) = 163 пм, РРОР = 134 °]. Длины остальных связей фосфора с кислородом лежат в пределах 145 ё 148 пм.
Для пирофосфорной кислоты характерны соли двух типов: кислые М2Н2Р2О7 и средние М4Р2О7. Первые, как правило, хорошо растворимы в воде, причём растворы их показывают кислую реакцию (в 1%-ном растворе Na2H2P2O7 рН = 4,2). Из вторых растворимы только соли наиболее активных одновалентных металлов. Растворы их имеют щелочную реакцию (в 1%-ном растворе Na4P2O7 рН = 10,2).
При нагревании пирофосфорной кислоты до 300 °С постепенно образуется метафосфорная кислота. Она является полимерным соединением состава (НРО3)n и представляет собой бесцветную стекловидную массу, которая плавится около 40 °С. Метафосфорная кислота (главным образом Н4Р4О12) получается также при взаимодействии Р2О5 с малым количеством воды. В растворе она очень медленно (быстрее при кипячении и в присутствии сильных кислот) присоединяет воду и переходит в ортогидрат. Кислотные свойства (НРО3)n выражены очень сильно (последние константы диссоциации Н3Р3О9 и Н4Р4О12 равны соответственно К3 = 2·10-2 и К4 = 3·10-3). Из средних метафосфатов растворимы только соли Mg и наиболее активных одновалентных металлов. Остальные почти нерастворимы в воде, но растворяются в НNO3 или избытке НРО3 и её растворимых солей. Сплавлением NaH2PO4 с Н3РО4 могут быть получены кислые метафосфаты общей формулы NaxHy(PO3)x+y.
Некоторые соли отдельных метафосфорных кислот (с определёнными значениями n) были выделены в индивидуальном состоянии. Так, медленным взаимодействием фосфорного ангидрида с раствором соды на холоду может быть получен Nа4Р4О12·4Н2О. Для триметафосфата натрия (Na3Р3О9) известны кристаллогидраты с 6 и 1, для гексаметафосфата (Nа6Р6О18) — с 6 молекулами воды.
Практически важен гексаметафосфат натрия, который может быть получен нагреванием NаН2РО4 до 700 °С (с последующим быстрым охлаждением расплава). Процесс его образования проходит, по-видимому, через следующие стадии:
160 250 525 650°С
NaH2PO4® Na2H2P2O7® (NaPO3)x® (NaPO3)3® (NaPO3)6
Гексаметафосфат натрия (т. пл. 610 °С) гигроскопичен и при хранении на воздухе расплывается, постепенно переходя в пирофосфат и затем в ортофосфат. В воде он труднорастворим. Раствор имеет слабокислую реакцию (рН » 6,5) и настолько прочно связывает катионы двухвалентных металлов (путём обменного разложения с образованием Na4ЭР6О16 или Nа2Э2Р6О18), что в нём медленно растворяется даже BaSO4. Гексаметафосфат натрия используют для умягчения воды и удаления накипи из паровых котлов, а также для предупреждения коррозии металлов.
Следует отметить, что вопрос о составе и строении метафосфатов ещё далеко не ясен. Возможно, что некоторые из описанных соединений этого типа представляют собой смеси веществ. Вместе с тем весьма вероятно существование метафосфатов с n » 6. В частности, обычный “гексаметафосфат” натрия, по-видимому, правильнее описывается формулой (NаРО3)n·Н2О, где n тем больше, чем выше применяемая при получении температура и меньше давление водяного пара в окружающей атмосфере. Что касается строения, то для низших членов, включая индивидуальные гексаметафосфаты, оно кольцевое — из связанных общими атомами кислорода тетраэдров РО4. В связях Р-О-Р ядерное расстояние d(PO) = 161 пм, в остальных — 149 пм. Более высокомолекулярные метафосфорные кислоты строятся по типу (НО)2ОР-··· -ОР(О)(ОН)- ··· -ОРО(ОН)2 с двумя гидроксильными группами на концах более или менее длинной цепи из радикалов НРО3. Схемы координации тетраэдров РО4 в таких цепях могут быть различными. Кислотный характер водородов цепи выражен сильнее, чем концевых.
При сильном накаливании метафосфаты с отщеплением Р2О5 переходят в пиро- и затем в ортофосфаты, например, по уравнениям:
2 Са(РО3)2 = Р2О5 + Са2Р2О7 (>900 °С) и
3 Са2Р2О7 = Р2О5 + 2 Са3(РО4)2 (>1200 °С).
Деполимеризация полифосфатов щелочных металлов может быть вызвана их сплавлением при 700 °С с перхлоратами (процесс сопровождается частичным выделением хлора и кислорода).
Для общей характеристики фосфатов была предложена схема, основанная на величине мольного отношения (М2О + Н2О)/Р2О5, где М — эквивалент металла. Как видно из рис. 2, термин “метафосфаты” отнесён в ней лишь к соединениям стехиометрического состава.
(М2О + Н2О)/Р2О5
0 1 2 3
![]()
![]()
![]()
![]()
![]() Ѕ Ѕ Ѕ Ѕ
Ѕ Ѕ Ѕ Ѕ
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Ультрафосфаты Полифосфаты Орто + пиро- Ортофосфаты
фосфаты и двойные соли
Метафосфаты Пирофосфаты Ортофосфаты
Рис. 2. Схема общей классификации фосфатов.
При нагревании полифосфаты хорошо сцепляются с металлами и сообщают огнеупорность их поверхностям. Полифосфаты натрия являются обычными составными частями стиральных порошков. Практическое значение для качественного химического анализа имеет образование метафосфорнокислого натрия при прокаливании так называемой “фосфорной соли”:
NaNH4HPO4 = NaPO3 + NH3 + H2O.
Расплавленный метафосфат натрия легко реагирует с оксидами металлов, образуя соответствующие ортофосфаты, например, по уравнениям:
NaPO3 + CoO = NaCoPO4 или
3 NaPO3 + Cr2O3 = 2 CrPO4 + Na3PO4.
Так как получающиеся фосфаты часто бывают окрашены в характерные цвета (например, Со — в синий, Сr — в зелёный), образованием их иногда пользуются для открытия соответствующих металлов.
Для отличия ортофосфорной кислоты от мета- и пирофосфорной пользуются реакцией их солей с АgNO3, образующим в присутствии иона РО4’’’жёлтый осадок Аg3PO4, а в присутствии ионов Р2О7’’’’ и РО3’ — белый осадок соответствующей серебряной соли. Две последних кислоты отличаются друг от друга по их разному действию на белок: пирофосфорная его не свёртывает, метафосфорная свёртывает.
Сплавлением смеси NaH2PO4+2Na2HPO4 может быть получена соль состава Na5P3O19, являющаяся производным не выделенной в индивидуальном состоянии трифосфорной кислоты Н5Р3О10. В 1%-ном растворе Na5P3O10 pH = 10,0. Ион Р3О105- образован тремя тетраэдрами РО4, из которых средний имеет по одному общему атому кислорода с двумя другими. Свыше 620 °С он распадается на пирофосфат и метафосфат натрия (из которых вновь образуется при медленном охлаждении системы). В растворах соль эта при обычных условиях довольно устойчива и лишь медленно (гораздо быстрее при подкислении) гидролизуется до ортофосфата.
При высоких концентрациях фосфорного ангидрида в системе Р2О5-Н2О имеют место сложные равновесия между различными кислотами фосфора. В расплавленном (или стеклообразном) состоянии ни одна из кислот не является индивидуальным химическим соединением. По другим данным, рассматриваемая система состоит из смеси Н3РО4 с различными линейно полимеризованными фосфорными кислотами, имеющими в молекуле до 10 и даже более атомов фосфора. Кипящая при 869 °С азеотропная смесь фосфорного ангидрида с водой содержит 92% Р2О5 и приблизительно отвечает составу 3Р2О5·2Н2О.
Хотя гидрат фосфорного ангидрида типа Н7РО6 (т.е. Р2О5 + 7Н2О) неизвестен, однако могут быть получены его производные, в которых кислороды замещены на кислотные остатки некоторых других кислот, в частности на МоО42-, Мо2О72-, WO42-, W2O72-. Комплексные кислоты подобного типа называются гетерополикислотами. Практическое значение из этих производных имеет кислый молибдофосфат аммония (NH4)3H4[P(Mo2O7)6]. Образованием этой труднорастворимой интенсивно жёлтой соли пользуются для открытия Н3РО4. Реакция идёт по уравнению:
H3PO4 + 12 (NH4)2MoO4 + 21 HNO3 = (NH4)3H4[P(Mo2O7)6]Ї + 21 NH4NO3 + 10 H2O.
Производные фосфорной кислоты находят многообразное применение в различных отраслях промышленности. Однако особенно велико их значение для сельского хозяйства. В частности, это относится к кислому фосфату кальция состава Са(Н2РО4)2·Н2О, который является основой важнейшего фосфорсодержащего минерального удобрения — суперфосфата.
Суперфосфат получают обработкой предварительно размолотых природных фосфоритов (или апатитовых концентратов) серной кислотой. После тщательного перемешивания влажная масса некоторое время “вызревает”. При этом по схеме:
Са3(РО4)2 + 2 Н2SO4 = 2 CaSO4 + Ca(H2PO4)2
образуется смесь сульфата и дигидрофосфата кальция, которая после измельчения и применяется в качестве удобрения (под названием простого суперфосфата). Входящий в состав легкорастворимого Са(Н2РО4)2 фосфор хорошо усваивается растениями.
Большим недостатком рассматриваемого удобрения является наличие в нём бесполезного “балласта” в виде СаSO4. Для получения двойного суперфосфата из природного фосфорита сначала выделяют фосфорную кислоту по реакции:
Са3(РО4)2 + 3 Н2SO4 = 3 CaSO4Ї + 2 H3PO4.
Затем отделив осадок СаSO4, полученной кислотой обрабатывают новую порцию фосфорита:
Са3(РО4)2 + 4 Н3РО4 = 3 Са(Н2РО4)2.
Иногда вместо этого нейтрализуют Н3РО4 гидроксидом кальция, причём осаждается преципитат (СаНРО4·2Н2О), также являющийся хорошим удобрением. На многих почвах (имеющих кислый характер) фосфор довольно хорошо усваивается растениями непосредственно из тонко размолотого фосфорита (фосфорной муки).
Для дальнейшего концентрирования необходимых растениям элементов большое значение приобретает выработка смешанных удобрений. Важнейшим из них является аммофос [смесь NH4H2PO4 и (NH4)2HPO4], получаемый прямым взаимодействием аммиака и фосфорной кислоты. Тонна аммофоса заменяет три тонны простого суперфосфата и одну тонну (NH4)2SO4. Особенно удобна для пользования смесь аммофоса с солями калия (азофоска), содержащая все наиболее нужные растениям “удобрительные”— N, P и К. Соотношение между ними можно изменять в соответствии с особенностями почв и культур.
Галогенидные соединения фосфора образуются путём взаимодействия элементов, протекающего, как правило, весьма энергично. Галогениды типов РГ3 и РГ5 (кроме РI5) известны для всех галогенов, однако практическое значение из них имеют почти исключительно производные хлора.
Трёххлористый фосфор (РСl3) образуется при действии сухого хлора на взятый в избытке фосфор. Последний при этом воспламеняется и сгорает бледно-жёлтым пламенем по реакции:
2 Р + 3 Сl2 = 2 РСl3 + 665 кДж.
Трёххлористый фосфор представляет собой бесцветную жидкость, которая во влажном воздухе сильно дымит, а с водой энергично взаимодействует по уравнению:
РCl3 + 3 H2O = H3PO4 + 3 HCl.
Таким образом, РСl3 является хлорангидридом фосфористой кислоты.
При действии на РСl3 избытка хлора образуются бесцветные кристаллы гемипентахлорида фосфора:
РСl3 + Cl2Ы РСl5 + 130 кДж.
Как видно из уравнения, реакция эта обратима. При обычных условиях равновесие её смещено вправо, выше 300 °С — влево.
Будучи хлорангидридом фосфорной кислоты, РCl5 нацело разлагается водой, образуя Н3РО4 и НСl. Реакция проходит в две стадии (вторая медленнее первой):
РСl5 + H2O = POCl3 + HСl и POCl3 + 3 H2O = H3PO4 + 3 HCl.
Получающийся по первой из них хлороксид фосфора РОСl3 представляет собой бесцветную жидкость. Подобно обоим хлоридам фосфора, он применяется при органических синтезах. Пары всех трёх соединений ядовиты.
Пространственное строение галогенидов РГ3 отвечает треугольным пирамидам с атомом Р в вершине.
| Вещество | Теплота образования кДж/моль | Энергия связи Р-Г, кДж/моль | Агрегатное состояние при обычных условиях | Температура плавления, °С | Температура кипения, °С |
РF3 | 957 | 502 | бесцв. газ | -151 | -101 |
PCl3 | 313 | 322 | бесцв. жидкость | -90 | +75 |
PBr3 | 176 | 263 | бесцв. жидкость | -40 | +173 |
PI3 | 46 | 184 | красные кристаллы | +61 | разл. |
Очень ядовитый РF3 способен функционировать в качестве донора преимущественно по отношению к переходным металлам. Так, для них известны комплексы Э[PF3]6 (где Э — Сr, Mo, W), [Э(PF3)5]2 (где Э — Mn, Re), а также ГRe(PF3)5. Из приведённых формул вытекают значности металлов (0, ±1), совершенно не характерные для них в обычных соединениях.
Как правило, эти и им подобные продукты присоединения PF3 представляют собой бесцветные более или менее летучие в вакууме кристаллы, нерастворимые в воде, но растворяющиеся в органических растворителях, устойчивые к разбавленным кислотам, но легко разлагаемые щелочами. Получают их различными методами. Например, комплекс вольфрама синтезируют при 250°С путём взаимодействия WCl6 с порошком меди под давлением PF3 в 250 атмосфер. Легко возгоняющиеся в высоком вакууме уже около 40 °С кристаллы W(PF3)6 плавятся при 214 °С, а начинают разлагаться лишь выше 320 °С.
Жидкие галогениды РГ3 растворяют белый фосфор без химического взаимодействия с ним. Вместе с тем известны фторид, хлорид и иодид общего типа Г2Р-РГ2 с транс-строением молекул. Первое из этих соединений было получено по реакции:
2 РF2I + 2 Hg = Hg2I2 + P2F4
и при обычных условиях газообразно (т. пл. -86, т. кип. -6 °С). Молекула F2P-PF2 частично распадается на радикалы РF2. Под действием НI она легко расщепляется на РF2I и PF2H.
Хлорид (Р2Сl4) образуется при действии тихого электрического разряда на смесь паров РСl3 с водородом. Он представляет собой бесцветную жидкость (т. пл. -28 °С, т. кип. 180 °С), медленно разлагающуюся уже при обычных условиях.
Соответствующий иодид (Р2I4) может быть получен непосредственно из элементов. Он образует оранжевые кристаллы (т. пл. 126 °С), слагающиеся из полярных молекул.
Теплоты образования из элементов галогенидов РГ5 быстро уменьшается по ряду (кДж/моль) 1593 (F), 435 (Cl), 230 (Br), а йодистое производное неизвестно. Энергии связей равны 460 (РF), 260 (PСl) и 210 (PBr) кДж/моль. Пятифтористый фосфор представляет собой бесцветный газ (т. пл. -94, т. кип. -85 °С), пятихлористый — летучее твёрдое вещество (т. возг. 159, т. пл. 160 °С под давл.), а пятибромистый (т. пл. 106 °С с разл.) известен в двух формах: красной и светло-жёлтой.
В парах РВr5 полностью диссоциирован на РВr3 и Br2, а молекулы двух других пентагалогенидов имеют строение типа тригональной бипирамиды. Решётка кристаллического РСl5 состоит из ионов [PСl4]+ и [PСl6]-, а кристаллического РВr5 — из ионов [PВr4]+ и Br-.
РСl3 может быть окислен кислородом воздуха до РОСl3 (т. пл. +1, т. кип. 107 °С). При обычных условиях реакция идёт крайне медленно, но может быть значительно ускорена, если кислород заменить озоном или проводить её в присутствии нагретого катализатора (платиновой черни). Ещё более энергично, со взрывом, протекает образование газообразного при обычных условиях РОF3 (т. пл. -39 °С (под давл.), т. возг. -40 °С), если дать реакции первоначальный толчок пропусканием сквозь смесь РF3 и О2 электрической искры.
Для химии фосфора важна реакция по схеме:
НF + PF5Ы HPF6.
В подходящей среде при низких температурах (например в жидкой SO2 при -20 °С) процесс этот протекает слева направо и ведёт к образованию гексафторофосфорной кислоты (НРF6). Последняя представляет собой бесцветную маслянистую жидкость, при обычных температурах постепенно разлагающуюся на исходные вещества.
Расплавленный белый фосфор хорошо растворяет серу, но химическое взаимодействие между обоими элементами наступает лишь при достаточном нагревании их смеси (или сероуглеродного раствора). Важнейшими и лучше изученными из них являются Р4S3, P4S7 и P4S10. Путём перекристаллизации (например, из расплавленного нафталина) все три сульфида могут быть выделены в виде хорошо образованных жёлтых кристаллов.
По стороению молекула Р4S10 подобна молекуле Р4О10. Точки кипения рассматриваемых сульфидов лежат высоко (соответственно при 408, 523 и 514 °С), при нагревании в отсутствие воздуха они кипят без разложения, причём плотности двух первых соединений отвечают приведённым выше формулам, а последнего формуле Р2S5.
Взаимодействие галогенидов фосфора с избытком жидкого аммиака ведёт к первичному образованию амидов Р(NH2)3 или P(NH2)5, дальнейшие превращения которых описываются следующими схемами:
Р(NH3)3-NH3® HNPNH2-NH3® P2(NH)2-H2® P4N6-N2® PN
P(NH2)5-NH3® HNP(NH2)3-NH3® NP(NH2)2-NH3® NPNH -NH3® P3N5-N2® PN
Амиды фосфора крайне неустойчивы и не получены; малоустойчивы также продукты отщепления от них по одной молекуле аммиака — амидоимиды НNPNH3 и HNP(NH2)3. Напротив, остальные азотные производные фосфора термически более или менее устойчивы. Так, отщепление аммиака от NP(NH2)2 происходит лишь при нагревании выше 125 °С в вакууме. Образующийся NPNH (фосфам) представляет собой лёгкий белый порошок, нерастворимый в воде, щелочах и кислотах.
Накаливание фосфама в вакууме выше 400 °С сопровождается отщеплением аммиака с образованием нитрида пятивалентного фосфора — P3N5. Дальнейшее прокаливание последнего до 700 °С и выше ведёт к распаду его на жёлтый (при 700 °С) или красно-коричневый (выше 700 °С) нитрид трёхвалентного фосфора РN и свободный азот. Лишь выше 700 °С переходит в PN и бесцветный смешанный нитрид трёх- и пятивалентного фосфора — Р4N6 (следует отметить, что химическая индивидуальность этого аморфного и самовоспламеняющегося на воздухе вещества не бесспорна). Выше 800 °С PN распадается на элементы.
Подобно азоту, фосфор проходит в природе определённый цикл превращений. При образовании земной коры часть фосфора была, вероятно, связана металлами, причём получившиеся фосфиды вошли в состав более глубоких слоёв земной оболочки. Другая часть соединилась с кислородом в Р2О5. Этот кислотный ангидрид, комбинируясь с оксидами металлов, образовал затем ряд минералов, в большинство которых наряду с Р2О5 оказались включёнными и другие кислотные оксиды. Подобные фосфорнокислые или смешанные минералы в последующие геологические эпохи постепенно разлагались под действием воды и углекислого газа с частичным выделением растворимых солей фосфорной кислоты.
Последние, вероятно, играли значительную роль при возникновении простейших живых организмов. Дальнейшее развитие на Земле растительного покрова повело к извлечению фосфорнокислых солей из почвы с переводом их в сложные фосфорсодержащие белковые вещества, которые с растительной пищей попадали затем в организмы животных и подвергались там дальнейшей переработке. После отмирания животных и растений их останки попадали в почву, где фосфорсодержащие соединения постепенно распадались с образованием в конечном счёте солей фосфорной кислоты. Таким путём весь круговорот фосфора в природе может быть выражен простой суммарной схемой:
Рпочвы Ы Рбелка.
Почва, следовательно, получает обратно столько же фосфора, сколько было из него взято. Так как фосфорнокислые соли прочно удерживаются ею и почти не вымываются водой, содержание фосфора на том или ином участке земной поверхности при свободном протекании природных процессов с течением времени либо не изменяется, либо изменяется лишь незначительно.
Существенную поправку в этот баланс вносит сознательная деятельность человека. Культурные растения при своём произрастании извлекают из почвы определённые количества фосфора (кг на тонну):
| Озимая рожь | Яровая пшеница | Картофель | Сахарная свекла | ||||
| зерно | солома | зерно | солома | клубни | ботва | корни | ботва |
| 3,7 | 1,1 | 3,7 | 0,9 | 0,7 | 0,7 | 0,4 | 0,4 |
В результате урожаи всего мира ежегодно уносят с полей около 10 млн. т фосфора. Так как природных источников пополнения почвы его соединениями почти не существует, постепенно развивающийся в ней “фосфорный голод” проявляется более остро, чем азотный.
Седьмая группа периодической системы.
Из членов данной группы водород был рассмотрен ранее. Непосредственно следующие за ним элементы — F, Сl, Br и I — носят общее название галогенов. К ним же следует отнести и элемент № 85 — астат (Аt). Другую часть группы составляют элементы подгруппы марганца (Мп, Тс, Rе).
Как видно из приводимых электронных структур, атомы галогенов имеют 7 электронов во внешнем слое. Основываясь на этом, можно наметить некоторые черты их химической характеристики: так как до устойчивой конфигурации внешнего слоя не хватает лишь по одному электрону, наиболее типичным для галогенов должны быть соединения, в которых эти элементы играют роль одновалентных неметаллов. С другой стороны, их максимальную положительную валентность можно ожидать равной семи.
Иначе обстоит дела в подгруппе марганца. Здесь незаконченными являются уже два внешних слоя. Так как в наиболее удаленном от ядра слое находится только два электрона, тенденции к дальнейшему присоединению электронов не будет. Наоборот, при их отдаче и образовании валентных связей могут принять участие и 5 электронов следующего слоя. Поэтому максимальную положительную валентность элементов подгруппы марганца также можно ожидать равной семи. Таким образом, по своим основным тенденциям элементы обеих групп сильно отличаются друг от друга: тогда как галоиды должны в первую очередь характеризоваться резко выраженной металлоидностью, марганец и его аналоги будут вести себя как металлы.
Фтор.
На земной поверхности фтор встречается исключительно в составе солей. Общее его содержание в земной коре равняется 0,02 %. Основная масса фтора распылена по различным горным породам. Из отдельных форм его природных скоплений наиболее важен минерал флюорит — СаF2.
Фтор является “чистым элементом” — состоит только из атомов 19F. Впервые он был обнаружен в плавиковой кислоте (1810 г.). Попытки выделить этот элемент долгое время оставались безуспешными, и свободный фтор удалось получить лишь в 1886 г.
Основная масса фтора земной поверхности обязана своим происхождением горячим недрам Земли (откуда этот элемент выделяется вместе с парами воды в виде НF). Среднее содержание фтора в почвах составляет 0,02 %, в водах рек — 2·10-5 % и в океане — 1·10-4 %. Человеческий организм содержит фтористые соединения главным образом в зубах и костях. В вещество зубов входит около 0,01 % фтора, причем большая часть этого количества падает на эмаль [состав которой близок к формуле Са5F(РO4)3]. В отдельных костях содержание фтора сильно колеблется. Для растительных организмов накопление фтора не характерно. Из культурных растений относительно богаты им лук и чечевица. Обычное поступление фтора в организм с пищей составляет около 1 мг за сутки.
Установлено, что содержание фтора в питьевой воде сильно влияет на состояние зубов людей (и животных). Наилучшим является наличие около 1 мг фтора в литре. Содержание ниже 0,5 мг/л способствует развитию кариеса, а выше 1,2 мг/л — крапчатости эмали. В обоих случаях зубы подвергаются более или менее быстрому разрушению.
Элементарный фтор получают путем электролиза фтористых соединений. причем он выделяется на аноде по схеме:
2 F-® 2 е-+ 2 F ® 2 е + F2
Электролитом обычно служит смесь состава КF·2НF (часто с добавкой LiF). Процесс проводят при температурах около 100 'С в стальных электролизерах со стальными катодами и угольными анодами.
Удобная лабораторная установка для получения фтора показана на рис.
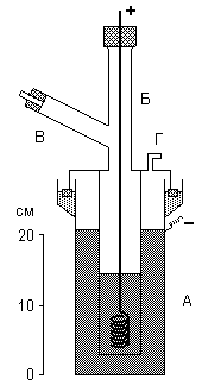
Рис.. Электролизёр для получения фтора.
Электролизу подвергают легкоплавкую смесь состава КF·3НF, помещенную в служащую катодом внешний медный сосуд А. Анод из толстой никелевой проволоки помещается в медном цилиндре Б, нижняя боковая часть которого имеет отверстия. Выделяющийся фтор отводится по трубке В (а водород — через отвод Г). Все места соединения отдельных частей прибора делают на пробках из СаF2 и замазке из РbО и глицерина.
Свободный фтор состоит из двухатомных молекул и представляет собой почти бесцветный (в толстых слоях зеленовато -желтый) газ с резким запахом. Он сгущается в светло-желтую жидкость при -188 °С и затвердевает при -220 °С.
Критическая температура фтора равна -129 °С, критическое давление 55 атм. При температуре кипения жидкий фтор имеет плотность 1,5 г/см3, а теплота его испарения составляет 1,6 кДж/моль. Жидкий фтор, как и его смесь с жидким кислородом (“флокс”), может служить энергичным окислителем реактивных топлив. С SiО2 или стеклом фтор не реагирует. При охлаждении ниже -252 °С его желтоватые кристаллы обесцвечиваются.
С химической стороны фтор может быть охарактеризован как одновалентный неметалл, и притом самый активный из всех неметаллов. Обусловлено это рядом причин, в том числе легкостью распада молекулы F2 на отдельные атомы — необходимая для этого энергия составляет лишь 159 кДж/моль (против 493 кДж/моль для О2 и 242 кДж/моль для С12). Атомы фтора обладают значительным сродством к электрону и сравнительно малыми размерами. Поэтому их валентные связи с атомами других элементов оказываются прочнее аналогичных связей прочих металлоидов (например, энергия связи Н-F составляет — 564 кДж/моль против 460 кДж/моль для связи Н-О и 431 кДж/моль для связи Н-С1).
Связь F-F характеризуется ядерным расстоянием 1,42 А. Для термической диссоциации фтора расчетным путем были получены следующие данные:
Температура, °С | 300 | 500 | 700 | 900 | 1100 | 1300 | 1500 | 1700 |
| Степень диссоциации, % | 5·10-3 | 0,3 | 4,2 | 22 | 60 | 88 | 97 | 99 |
Атом фтора имеет в основном состоянии структуру внешнего электронного слоя 2s22p5 и одновалентен. Связанное с переводом одного 2р-элсктрона на уровень 3s возбуждение трехвалентного состояния требует затраты 1225 кДж/моль и практически не реализуется.
Сродство нейтрального атома фтора к электрону оценивается в 339 кДж/моль. Ион F- характеризуется эффективным радиусом 1,33 А и энергией гидратации 485 кДж/моль. Для ковалентного радиуса фтора обычно принимается значение 71 пм (т. е. половина межъядерного расстояния в молекуле F2).
Подавляющее большинство м е т а л л о в соединяется с фтором уже при обычных условиях. Однако взаимодействие часто ограничивается образованием плотной поверхностной пленки фтористого соединения, которая предохраняет металл от дальнейшего разъедания. Так ведут себя, например, Сr, Ni и Мg, которые поэтому оказываются практически устойчивыми по отношению к фтору (в отсутствие воды).
Так как фтористые производные м е т а л л о и д н ы х элементов обычно легколетучи образование их не предохраняет поверхность металлоида от дальнейшего действия фтора. Поэтому взаимодействие часто протекает значительно энергичнее, чем со многими металлами. Например, кремний, фосфор и сера воспламеняются в газообразном фторе. Аналогично ведет себя аморфный углерод (древесный уголь), тогда как графит реагирует лишь при температуре красного каления. С азотом и кислородом фтор непосредственно не соединяется.
От водородных соединений других элементов фтор отнимает водород. Большинство оксидов разлагается им с вытеснением кислорода. В частности, вода взаимодействует по схеме
F2 + Н2О ® 2 НF + O
причем вытесняемые атомы кислорода соединяются не только друг с другом, но частично также с молекулами воды и фтора. Поэтому, помимо газообразного кислорода, при этой реакции всегда образуются пероксид водорода и оксид фтора (F2О). Последняя представляет собой бледно-желтый газ, похожий по запаху на озон.
Окись фтора (иначе — фтористый кислород — ОF2) может быть получена пропусканием фтора в 0,5 н. раствор NаОН. Реакция идет по уравнению:
2 F2 + 2 NаОН = 2 NаF + Н2О + F2О
Молекула F2О имеет структуру равнобедренного треугольника [d(FО) = 141 пм РFОF = 103°] и небольшой дипольный момент (m = 0,30), Для средней энергии связи O-Г дается значение 192 кДж/моль.
При охлаждении до -145 °С окcид фтора сгущается в желтую жидкость (плотность 1,5 г/см3), затвердевающую при -224 °С. Критическая температура F2О равна -58 °С, критическое давление 49 атм. Жидкая оксид фтора смешивается в любых соотношениях с жидкими O2, F2, O3 и способен растворять большие количества воздуха. Несмотря на эндотермичность F2О (теплота образования 25 кДж/моль), он все же сравнительно устойчив, например еще не разлагается при нагревании до 200 °С (энергия активации термического разложения равна 171 кДж/моль). Почти неразлагается оксид фтора и холодной водой, в которой он малорастворим (7: 100 по объему при 0 °С). Напротив, в щелочной среде (или под действием восстановителей) разложение F2О идет довольно быстро. Смесь его с водяным паром при нагревании взрывается реакция идет по уравнению:
ОF2 + Н2О = 2 НF + O2 + 326 кДж
Оксид фтора является сильным окислителем и очень ядовит.
Формально отвечающая F2О, как ангидриду, фторноватистая кислота (НОF) частично образуется при взаимодействии медленного тока фтора под уменьшенным давлением с охлаждаемой водой. Выделенная лишь в очень малых количествах (порядка мг), она представляет собой бесцветное вещество (т. пл. — 117 °С) с высоким давлением пара (5 мм рт. ст. уже при — 64 °С), в обычных условиях довольно быстро разлагающееся на НF и O2. Молекула НОF характеризуется следующими параметрами: d(ОН) = 0,96, d(ОF) = 1,44 А, РНОF = 97°. Фторноватистая кислота является, по-видимому, сильной, но водой она быстро гидролизуется, в основном по уравнению:
НОF + НОН = НF + Н2О2
Соли ее не получены, но известны вещества, которые можно рассматривать как продукты замещения ее водорода на радикалы металлоидного характера, т. е. как г и п о ф т о р и т ы этих радикалов.
Подобно самому фтору, его окись является одним из возможных эффективных окислителей реактивных топлив. Например, при использовании углеводородов СnН2n в качестве горючего и жидких F2 и F2О в качестве окислителя относительные (O2 = 1) расчетные значения удельного импульса и скорости ракет составляют 1,05 и 1,2 для F2, или 1,16 и 1,4 для F2О.
При действии тихого электрического разряда на охлаждаемую жидким воздухом смесь газообразных фтора и кислорода образуются оранжево-красные кристаллы состава F2O2 (т. пл. -163 °С). Оксид этот (дифтордиоксид) устойчив лишь ниже -80 °С, а при дальнейшем нагревании начинает распадаться на элементы. Изучавшееся при очень низких температурах взаимодействие его с различными другими веществами протекает, как правило, чрезвычайно бурно (нередко — со взрывом).
Молекула F2О2 полярна (m = 1,44) и по строению подобна молекуле перекиси водорода. Она характеризуется параметрами d(FО) = 158 пм, d(OO) = 122 пм, РООГ = 110° при угле около 88° между связями F-O. Так как d(OO) в молекуле O2 равно 121 пм, можно думать, что присоединение к ней двух атомов фтора существенно не искажает ее внутреннюю структуру (т. е. что дифтордиоксиду отвечает формула F-O-O-F с четырехвалентными атомами кислорода). Энергии связей OO и ОF оцениваются соответственно в 564 и 75 кДж/моль.
Гомологами F2О и F2О2 являются окислы фтора общей формулы F2Оn, где n = 3, 4, 5, 6. Они были получены действием тихого электрического разряда на смеси фтора с кислородом при температурах порядка -200 °С и под сильно уменьшенным давлением (например, синтез F2О6 велся при -210 °С и давлении около 1 мм рт. ст.). Все эти полипероксиды фтора представляют собой жидкие или твердые коричнево-красные вещества, устойчивые лишь при очень низких температурах (например, F2О6. — ниже -200 °С) и являющиеся чрезвычайно сильными окислителями. Интересно, что F2O3 нерастворим в жидком O2 или F2 (отличие от F2O2).
Практическое использование свободного фтора развилось сравнительно недавно. Потребляется он главным образом для фторирования органических соединений (т. е. замены в них водорода на фтор).
Процесс этот приобрел большое значение, так как многие фторорганические производные обладают весьма ценными свойствами. Необходим фтор и для получения соединений инертных газов.
Наиболее интересными с общехимической точки зрения производными фтора являются фториды инертных газов. Лучше других изученные соединения ксенона могут быть получены из элементов при нагревании, под действием электрического разряда или ультрафиолетовых лучей. Фториды ксенона — ХеF2, ХеF4 и ХеF6 — представляют собой бесцветные легко возгоняющиеся кристаллические вещества
Интересно, что средняя энергия связи Хе-F в них практически одинакова (132,9ё127,9 кДж/моль). Они хорошо (XeF2, ХеF6) или умеренно (ХеF4) растворимы в жидком фтористом водороде, а по донорной способности располагаются в ряд: ХеF42 6. Водой фториды ксенона разлагаются. В процессе гидролиза обычно возникает желтая окраска, которая затем исчезает.
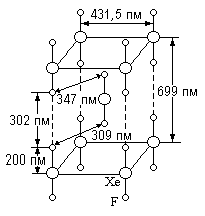
Рис.. Кристаллическая структура XeF2.
Ксенондифторид медленно образуется под действием дневного света на смесь Хе и F2 уже при обычных условиях (теплота образования 176 кДж/моль). Он обладает характерным тошнотворным запахом. Его давление пара составляет около 3 мм рт. ст. при обычных условиях и 760 мл рт. ст. при 155 °С. Теплота возгонки (сопровождающейся реакцией по схеме 2 ХеF2 = Хе + ХеF4) равна 29,4 кДж/моль. Строение кристалла XeF2 (т. пл. 129 °С) показано на рис.. Молекула ХеF2, линейна, связи Хе-F в ней характеризуются длиной 198 пм. По-видимому, они имеют сильно выраженный полярный характер. Для возможности образования этих связей необходимо возбуждение атома ксенона от его нормального нульвалентного состояния (5s25р6) до одного из ближайших двухвалентных, что требует значительной затраты энергии (803 кДж/моль при возбуждении до 5s25p56s1, 924 кДж/моль — до 5s25р56р1 или 953 кДж/моль — до 5s25р55d1). Растворимость ХеF2 в воде составляет около 0,15 моль/л при 0 °С. Раствор является сильнейшим окислителем — потенциал системы ХеF2-Хе в кислой среде равен 2,2 в. Саморазложение раствора по схеме2 ХеF2 + 2 Н2О = 4 НF + 2 Хе + O2
в кислой среде идет медленно, а в щелочной очень быстро.
Ксенонтетрафторид образуется из элементов с довольно значительным выделением тепла (251 кДж/моль) и является наиболее устойчивым из всех фторидов ксенона. Молекула его имеет структуру квадрата с атомом Хе в центре, а связь Хе-F характеризуется длиной 195 пм (в кристалле) или 185 пм (в газе) и полярностью. Давление пара составляет около 3 мм рт. ст. при обычных условиях и 760 мм рт. ст. при 146 °С, а теплота возгонки равна 15,3 кДж/моль. Ксенонтетрафторид образует с ХеF2 кристаллический аддукт ХeF2·ХеF4, но не взаимодействует с КF или ВF3. Ртуть он фторирует:
ХеF4 + 2 Нg = 2 НgF2 + Хе,
а раствор его в НF подобным же образом фторирует платину
ХеF4 + Рt = РtF4 +Хе
Иодистый калий (в растворе) количественно реагирует по уравнению:
4 KI + ХеF4 = 4 КF + 2 I2 + Хе
что находит аналитическое использование. Под действием воды ХеF4 разлагается по схеме 3 ХеIV = Хе0 + 2 ХеVI (в кислой среде) или 2 ХеIV = Хе0 + ХеVIII (в щелочной среде).
Был описан также о к с о ф т о р и д ХеОF2, образующийся (в качестве незначительной примеси) при нагревании сильно разбавленной кислородом или воздухом смеси Хе с F2. Для него даются следующие константы: точка плавления 90 °С и точка кипения — около 115 °С. Предполагается, что тот же состав имеет сконденсированный при -80 °С ярко-желтый продукт гидролиза ХеF4 водяным паром. Сообщалось, что это же вещество образовывалось, по-видимому, в результате взаимодействия Хе с большим избытком F2О2 при -118 °С. Однако существование ОХеF2 пока нельзя считать окончательно установленным.
Бесцветный ксенонгексафторид известен в трех различных кристаллических модификациях. Он плавится при 49 °С в желтую жидкость с низкой диэлектрической проницаемостью (e = 4,1 при 55 °С), по-видимому, содержащую тетрамерные ассоциаты. При затвердевании ХеF6 вновь обесцвечивается. Давление его пара (имеющего бледно-желтую окраску) составляет 30 мм рт. ст. при 25 °С и 760 мм рт. ст. при 76 °С. Ксенонгексафторид чрезвычайно химически активен и способен разлагаться со взрывом. Строение его молекулы пока точно не установлено, но известно, что он не обладает обычной для соединений типа ЭF6 симметрией правильного октаэдра. Среднее расстояние d(ХеF) = 190 пм.
Растворение ХеF6 в жидком фтористом водороде сопровождается частичной электролитической диссоциацией по схеме:
ХеF6 + НF = ХеF5++ НF2-
Насыщенный при обычных условиях раствор имеет состав, приблизительно отвечающий формуле ХеF6·6НF. В отличие от тетрафторида ХеF6 образует твердые продукты присоединения и с ВF3, и с фторидами щелочных металлов. Бесцветный Nа2ХеF8 разлагается ниже 100 °С, но Сs2ХеF8 — лишь выше 400 °С. Гораздо менее устойчивы соли типа МХеF7. Так, желтый СsХеF7 переходит в кремовый Сs2ХеF8 уже при 50 °С. Все эти соли чрезвычайно химически активны и бурно реагируют с водой (причем Хe сохраняется в растворе, по-видимому, как ХеО3).
Под действием влажного воздуха ксенонгексафторид частично гидролизуется с образованием о к с о ф т о р и д а ОХеF4. Последний представляет собой бесцветную жидкость (т. пл. -46, т. кип. 102 °С), менее реакционноспособную, чем ХeF6. Она смешивается с жидким фтористым водородом, а с фторидами тяжелых щелочных металлов образует следующие соединения: 3КF·ХеОF4, 3RbF·2ХеОF4, СsF·ХеОF4. Молекула ОХеF4 имеет m = 0,65 и структуру квадратной пирамиды с атомом Хe около середины основания из четырех атомов фтора [d(ХеО) = 1,70, d(ХеF) = 1,90 А, РОХеF = 92°].
Дальнейший медленный гидролиз ХеОF4 (или гидролиз ХеF4 в кислой среде с дисмутацией по схеме 3 Хе+4® Хе0+ 2 Хе+6) ведет к образованию к с е н о н т р и о к с и д a, который может быть выделен в виде крайне взрывчатых бесцветных кристаллов, расплывающихся на воздухе. Теплота образования ХеОз из элементов равна — 401 кДж/моль. В сухом состоянии это сильно эндотермичное соединение способно распадаться со взрывом, но при медленном нагревании выше 40 °С разложение на Хе и O2, идет спокойно (заканчиваясь при 140 °С). Молекула ХеОз имеет форму тригональной пирамиды с атомом Хе в вершине [d(ХеО) = 1,76 А, РОХеО = 103°]. Для средней энергии связи ХеО дается значение 117 кДж/моль.
Взаимодействием ХеО3 с ХеОF4 был получен ХеО2F2. Этот оксофторид представляет собой бесцветные кристаллы (т. пл. 31 °С). Во влажном воздухе он гидролизуется до ХeО3, а в сухом медленно разлагается на ХеF2 и O2. От ХеО3 производятся молекулярные соединения типа МF·ХеО3 (где М — Сs, Rb, К), а также СsС1·ХеО3 и СsВr·ХеО3. Строение их молекул отвечает, по-видимому, формуле М[ХеО3Г]. Фториды термически устойчивы до 200 °С.
Ксенонтриоксид хорошо растворим в воде, но лишь слабо взаимодействует с ней: равновесие по схеме
Н2О + ХеО3Ы Н2ХеО4Ы Н+ + НХеО4-
сильно смещено влево. При рН > 10,5 оно смещается вправо с образованием солей типа МНХеO4 или МН5ХеО6, (где M — Nаё Сs). Отвечающая этим к с е н а т а м кислота была получена при 0 °С взаимодействием ксенонтетрафторида с разбавленным раствором гидрооксида кальция по суммарному уравнению:
3 ХеF4 + 6 Са(ОН)2 = 6 СаF2Ї + Хе + 2 Н2ХеО6
При низких температурах (порядка -25 °С) она может сохраняться длительное время. Ее бариевая соль — Ва3ХеО6 — малорастворима в воде (0,25 г/л при 25 °С) и испытывает термический распад лишь при 125 °С. В сильнощелочной среде шестивалентный ксенон неустойчив (дисмутирует по схеме 4 Хе+6 = Хе0 + 3 Хе+8). Напротив, кислые водные растворы ХеО3 вполне устойчивы. Для окислительных потенциалов системы Хе+6-Хе0даются значения +2,1 в (в кислой среде) и +1,2 в (в щелочной среде).
При действии озона на раствор ХеО3 в 1 М NаОН образуется Nа4ХеО6. Анион этого п е р к с е н а т а имеет структуру слегка искаженного октаэдра со средним расстоянием d(ХеО) = 185 пм. Тетранатрийперксенат может быть выделен в виде бесцветного кристаллогидрата с 6 или 8 Н2О, который обезвоживается около 100 °С, а бурно разлагается лишь при 360 °С. Соль эта малорастворима в воде (растворимость около 0,025 М), но сильно гидролизуется, давая щелочную реакцию. Последнее обусловлено относительной слабостью ксеноновой кислоты, которой отвечают следующие значения последовательных констант диссоциации: К1 = 10-2, К2 = 10-6 и К3 = 3·10-11. Содержащие Хе+8 водные растворы постепенно отщепляют кислород, переходя в растворы Хе+6, причем скорость такого перехода возрастает с уменьшением рН среды (уже при рН = 7 он осуществляется почти мгновенно). Для окислительных потенциалов системы Хе+8-Хе+6 даются значения +2,3 в (в кислой среде) и +0,9 в (в щелочной среде). Смешанным производным этих валентностей является полученное озонированием смеси растворов ХеО3 и КОН взрывчатое желтое молекулярное соединение состава К4ХеО6·2ХеО3.
Взаимодействием Nа4ХеО4 с безводной Н2SO4 при низких температурах был получен желтый к с е н о н т е т р о к с и д (теплота образования из элементов -644 кДж/моль). Молекула ХеО4 имеет структуру тетраэдра с атомом ксенона в центре, а связь ХеО характеризуется ядерным расстоянием d(ХеО) = 174 пм и энергией 88 кДж/моль. Давление пара этого оксида составляет 3 мм рт. ст. при -35 °С. В твердом состоянии он уже ниже 0 °С медленно разлагается на Хе и О2, а в газообразном при комнатной температуре — на ХеО3, Хе и О2.
Сообщалось также об образовании при взаимодействии Nа4ХеО6 и ХеF6 очень летучего ХеО3F2, но выделен он не был.
Имеются отдельные указания на возможность образования в тех или иных условиях нестойкого соединения ксенона с хлором — ХеС12 или ХеС14. Однако все такие указания даются лишь предположительно, и считать, что хлориды ксенона существуют, пока нельзя.
Взаимодействие с фтором радона идет легче, чем ксенона (но состав фторидов не устанавливался), а криптона — гораздо труднее. Известен только к р и п т о н д и ф т о р и д, который был впервые получен действием электроразряда на смесь элементов при -188 °С. Он представляет собой бесцветные кристаллы, давление пара над которыми равно 30 мм рт. ст. при 0 °С, а теплота возгонки составляет 36,8 кДж/моль. Молекула КrF2 линейна, а связь КrF характеризуется ядерным расстоянием d(KrF) = 188 пм, энергией 50 кДж/моль. При низких температурах КrF2 может сохраняться неделями, а при 20 °С за час разлагается около 10 % исходного количества. Его насыщенный раствор в жидком фтористом водороде по составу приблизительно отвечает формуле КrF2·3НF. Получить какие-либо производные аргона (и еще более легких инертных газов) пока не удалось.
Как видно из изложенного выше, сведения о впервые полученных в 1962 г. соединениях инертных газов еще довольно отрывочны (и отчасти недостоверны). Однако сам факт существования этих соединений имеет большое принципиальное значение, так как наиболее наглядно и убедительно опровергает постулат незыблемости электронного октета. Тем самым ставится также вопрос о целесообразности отказа от уже не вполне отвечающего существу названия “инертные газы” (подходящей его заменой могло бы служить название а э р о ф и л ы). О широком практическом использовании соединений инертных газов говорить еще рано, но, например, устойчивый при обычных температурах ХеF4 мог бы служить, удобной реакционной формой фтора (не загрязненного никакими другими химически активными элементами). Следует лишь иметь в виду возможную взрывоопасность этого соединения (из-за образования взрывчатого ХеО3 во влажном воздухе).
В отличие от свободного фтора ф т о р и с т ы й в о д о р о д (НF) и многие его производные используются уже с давних пор.
Непосредственное соединение фтора с водородом сопровождается значительным выделением тепла:
Н2 + F2 = 2 НF + 543 кДж
Реакция протекает обычно со взрывом, который происходит даже при сильном охлаждении газов и в темноте. Практического значения для получения НF этот прямой синтез не имеет, но, в принципе, он может быть использован для создания реактивной тяги.
Промышленное получение фтористого водорода основано на взаимодействии СаF2 с концентрированной Н2SO4 по реакции:
СаF2 + Н2SO4 = СаSO4 +2 НF
Процесс проводят в стальных печах при 120-300 °С. Части установки, служащие для поглощения НF, делаются из свинца.
В качестве реактивного топлива смесь фтора с водородом способна давать удельный импульс 410 сек. Бесцветное пламя, возникающее при взаимодействии этих газов, может иметь температуру до 4500 °С. В лабораторных условиях для получения чистого фтористого водорода применяются обычно небольшие установки изготовленные целиком из платины (или меди). Исходным веществом служит тщательно высушенный бифторид калия (КF·НF), при нагревании разлагающийcя c отщеплением НF. Полученный продукт часто содержит примесь механически увлеченного бифторида. Для очистки его подвергают перегонке при 35-40 °С. Совершенно безводный или близкий к этому состоянию фтористый водород почти мгновенно обугливает фильтровальную бумагу. Этой пробой иногда пользуются для контроля степени его обезвоживания. Более точно такой контроль осуществляется определением электропроводности у безводного фтористого водорода она ничтожно мала, но даже следы воды (как и многих других примесей) резко ее повышают.
Фтористый водород (гидрофторид) представляет собой бесцветную, подвижную и легколетучую жидкость (т. кип. +19,5 °С), смешивающуюся с водой в любых соотношениях. Он обладает резким запахом, дымит на воздухе (вследствие образования с парами воды мелких капелек раствора) и сильно разъедает стенки дыхательных путей. Многие неорганические соединения хорошо растворимы в жидком НF, причем растворы являются, как правило, проводниками и электрического тока.
Связь Н-F характеризуется ядерным расстоянием 0,92 А. По отношению к нагреванию фтористый водород очень устойчив: его термическая диссоциация становится заметной лишь около 3500 °С.
Молекула НF весьма полярна (m = 1,74). С наличием на атомах значительных эффективных зарядов хорошо согласуется резко выраженная склонность фтороводорода к а с с о ц и а ц и и путем образования водородных связей по схеме ···Н-F···Н-F···.
Энергия такой связи составляет около 33,4 кДж/моль, т. е. она прочнее, чем водородная связь между молекулами воды.
Как показывает определение плотности пара, вблизи точки кипения молекулы газообразного фтористого водорода имеют средний состав, приблизительно выражаемый формулой (НF)4. При дальнейшем нагревании ассоциированные агрегаты постепенно распадаются и кажущийся (средний) молекулярный вес уменьшается, причем лишь около 90 °С достигает значения 20, соответствующего простой молекуле НF.
Критическая температура фтористого водорода равна 188 °С, критическое давление 64 атм. Теплота испарения жидкого НF в точке кипения составляет лишь 7,5 кДж/моль. Столь низкое значение (примерно в 6 раз меньшее, чем у воды при 20 °С) обусловлено тем, что само по себе испарение мало меняет характер ассоциации фтористого водорода (в отличие от воды).
Подобно плотности (0,99 г/см3), диэлектрическая проницаемость жидкого фтористого водорода (84 при 0 °С) очень близка к значению ее для воды. Существующая у жидкого фтористого водорода ничтожная электропроводность обусловлена его незначительной ионизацией по схеме:
НF + НF + НF Ы Н2F+ + НF2-
связанной с характерной для НF склонностью к образованию иона г и д р о д и ф т о р и д а — НF2- [имеющего линейную структуру с атомом водорода в центре и d(FF) = 227 пм]. Напротив, образование иона ф т о р о н и я (Н2F+) для НF нехарактерно, что и ограничивает самоионизацию (К = 2·10-11). Тенденция к образованию иона НF2-,, накладывает свой отпечаток на всю химию фтористого водорода.
Помимо воды, из неорганических соединений в жидком HF хорошо растворимы фториды, нитраты и сульфаты одновалентных металлов (и аммония), хуже — аналогичные соли Mg, Сa, Sr и Вa. По рядам Li-Сs и Мg-Ва, т. е. по мере усиления металлического характера элемента, растворимость повышается. Щелочные и щелочноземельные соли других галоидов растворяются в НF с выделением соответствующего галоидоводорода. Соли тяжелых металлов в жидком HF, как правило, нерастворимы. Наиболее интересным исключением является Т1F, растворимость которого очень велика (в весовом отношении около 6: 1 при 12 °С). Практически нерастворимы в жидком НF другие галоидоводороды. Концентрированная серная кислота взаимодействует с ним по схеме:
Н2SO4 + 3 НF Ы Н3О++ НSO3F + НF2-
Жидкий фтористый водород является лучшим из всех известных растворителем белков.
Растворы воды и солей в жидком фтористом водороде хорошо проводят электрический ток, что обусловлено диссоциацией, например, по схемам:
Н2О + 2 НF Ы Н3О++ НF2-
КNО3 + 2 НF Ы НNО3 + К+ + НF2-
НNО3 + 4 НF Ы Н3О+ + NО2+ + 2 НF2-
Аналогичное отношение к НF характерно и для многих кислородсодержащих органических молекул. Так, в водной среде глюкоза является типичным неэлектролитом, а в жидком НF наоборот, типичным электролитом за счет взаимодействия по схеме:
С6Н12O6 + 2 НF Ы [С6Н12О6·Н]+ + НF2-
Кристаллы твердого фтористого водорода слагаются из зигзагообразных цепей ···Н-F···Н-F···Н-F···, образованных при посредстве водородных связей. Расстояние d(FF) в таких цепях — 249 пм, а угол зигзага — 120°. Теплота плавления твердого НF (т. пл. -83 °С, плотность 1,6 г/см3) составляет 3,8 кДж/моль, что близко к значению для льда. Для жидкого фтористого водорода наиболее вероятно одновременное существование и цепей, и колец из молекул НF.
Химическая активность НF существенно зависит ~н отсутствия или наличия воды. Сухой фтористый водород не действует на большинство металлов. Не реагирует он и с оксидами металлов. Однако если реакция с оксидом начнется хотя бы в ничтожной степени, то дальше она некоторое время идет с самоускорением, так как в результате взаимодействия по схеме
МО + 2 НF = МF2 + Н2О
количество воды увеличивается.
Случаи взаимодействия сухого фтористого водорода с оксидами металлов и металлоидов, рассмотренные выше, могут служить типичным примером аутокаталитических реакций, т. е. таких процессов, при которых катализатор (в данном случае — вода) не вводится в систему извне, а является одним из продуктов реакции.
Как показывает рис. Ч11-4, скорость подобных процессов сначала, по мере увеличения в системе количества катализатора, нарастает до некоторого максимума, после чего начинает уменьшаться вследствие понижения концентраций реагирующих веществ.
***********РИС
Подобным же образом действует фтористый водород и на окислы некоторых металлоидов. Практически важно его взаимодействие с двуокисью кремния — SiO2 (песок, кварц), которая входит в состав стекла. Реакция идет по схеме
SiO2 + 4 НF = SiF4 + 2 Н2O
Поэтому фтористый водород нельзя получать и сохранять в стеклянных сосудах.
На взаимодействии НF и SiO2 основано применение фтористого водорода для травления стекла. При этом вследствие удаления частичек SiO2 его поверхность становится матовой, что и используют для нанесения на стекло различных меток, надписей и т. п.
Перед фигурным травлением стекла его обычно покрывают тонким слоем воска, а затем снимают этот слой на тех местах, которые должны быть протравлены. Под действием паров НF места эти становятся матовыми, тогда как под действием плавиковой кислоты они остаются прозрачными. Матовое травление в жидкости достигается предварительным добавлением к плавиковой кислоте нескольких процентов фтористого аммония.
В водном растворе НF ведет себя как одноосновная кислота средней силы. Продажный раствор этой фтористоводородной (иначе, п л а в и к о в о й) кислоты содержит обычно 40% НF.
Техническая плавиковая кислота обычно содержит ряд примесей — Fе, Рb, Аs, Н2SiF6, SO2) и др. Для грубой очистки ее подвергают перегонке в аппаратуре, изготовленной целиком из платины (или свинца), отбрасывая первые порции дистиллята. Если этой очистки недостаточно, то техническую кислоту переводят в бифторид калия, затем разлагают его нагреванием и растворяют получающийся фтористый водород в дистиллированной воде. Крепкая плавиковая кислота (более 60% НF) может сохраняться и транспортироваться в стальных емкостях. Для хранения плавиковой кислоты и работы с ней в лабораторных условиях наиболее удобны сосуды из некоторых органических пластмасс. Крупным потребителем фтористоводородной кислоты является алюминиевая промышленность.
Растворение фтористого водорода в воде сопровождается довольно значительным выделением тепла (59 кДж/моль). Характерно для него образование содержащей 38,3 % НF и кипящей при 112 °С азеотропной смеси (по другим данным 37,5 % и т. кип. 109 °С). Такая азеотропная смесь получается в конечном счете при перегонке как крепкой, так и разбавленной кислоты.
При низких температурах фтористый водород образует нестойкие соединения с водой состава Н2О·НF, Н2О·2НF и Н2О·4НF. Наиболее устойчиво из них первое (т. пл. -35 °С), которое следует рассматривать как фторид оксония — [Н3O]F.
Помимо обычной электролитической диссоциации по уравнению
HF Ы H+ + F- (К = 7·10-4),
для растворов фтористоводородной кислоты характерно равновесие:
F- + НF Ы НF2’
Значение константы этого равновесия ([НF2’]/[F’][НF]=5) показывает, что в не очень разбавленных растворах НF2’ содержится больше анионов чем простых анионов F’. Например, для приводимых ниже общих нормальностей (С) приближенно имеем:
С [НF] [Н+'] [F-] [HF2’]
0,100 0,088 (88 %) 0,009 (9 %) 0,006 (6 %) 0,003 (3 %)
1,000 0,890 (89 %) 0,006 (6 %) 0,010 (1 %) 0,050 (5 %)
Фтористоводородная кислота (ацидофторид) более или менее энергично реагирует с большинством металлов. Однако во многих случаях реакция протекает лишь на поверхности, после чего металл оказывается защищенным от дальнейшего действия кислоты слоем образовавшейся труднорастворимой соли. Так ведет себя, в частности, свинец, что и позволяет пользоваться им для изготовления частей аппаратуры, устойчивой к действию НF.
Соли фтористоводородной кислоты носят название ф т о р и с т ы х или ф т о р и д о в. Большинство их малорастворимо в воде — из производных наиболее обычных металлов хорошо растворяются лишь фториды Nа, К, Ag, A1, Sn и Нg. Все соли плавиковой кислоты ядовиты. Сама она при попадании на кожу вызывает образование болезненных и трудно заживающих ожогов (особенно под ногтями). Поэтому работать с плавиковой кислотой следует в резиновых перчатках.
Весьма характерно для фтористого водорода образование продуктов присоединения к фторидам наиболее активных металлов. Соединения эти, как правило, хорошо кристаллизуются и плавятся без разложения. Примером могут служить производные калия — КF·НF (т. пл. 239 °С), КF·2НF (62 °С), КF·3НF (66 °С) и КF·4НF (72 °С). Строение этих продуктов присоединения отвечает, вероятно, формулам вида К[F(НF)n] с водородными связями между ионом F- и молекулами HF. Разбавленные растворы гидродифторида калия (КНF2) применяются иногда для удаления пятен от ржавчины.
Работа с фтористым водородом и другими фторидами требует соблюдения мер предосторожности, так как все соединения фтора ядовиты. Сам фтористый водород, помимо резкого раздражения слизистых оболочек, вызывает также разрушение ногтей и зубов. Кроме того, он способствует осаждению кальция в тканях. Средство первой помощи при острых отравлениях фторидами служит 2 %-ный раствор СаС12. При ожогах плавиковой кислотой пораженное место следует длительно (несколько часов) промывать струей холодной воды, а затем наложить на него компресс из свежеприготовленной 20 %-ной взвеси MgО в глицерине.
Хроническое отравление фторидами может быть вызвано как повышенным их содержанием в питьевой воде, так и вдыханием их с воздухом в виде пыли. В результате подобного отравления наблюдается разрушение зубной эмали. Существенно увеличивается также хрупкость костей, что создает предпосылки для их переломов. Имеются указания на то, что повышенное содержание фторидов в воде и воздухе способствует заболеванию зобом. Помимо фторной промышленности, с возможностью хронического отравления фтористыми соединениями приходится особенно считаться при выработке алюминия и суперфосфата. Предельно допустимой концентрацией связанного фтора в воздухе производственных помещений считается 5·10-4 мг/л.
Практическое применение НF довольно разнообразно. Безводный используется главным образом при органических синтезах, а плавиковая кислота — для получения фторидов, травления стекла, удаления песка с металлического лития, при анализах минералов и т. д. Широкое применение находят также некоторые фториды которые будут рассмотрены при соответствующих элементах.
2. Хлор.
По распространенности в природе хлор близок к фтору на его долю приходится 0,02 % от общего числа атомов земной коры. Человеческий организм содержит 0,25 вес. % хлора.
Природный хлор состоит из смеси двух изотопов — 35Сl (75,5 %) и 37Сl (24,5 %). Он был впервые получен (действием МnО2 на соляную кислоту ) в 1774 г., но установление его элементарной природы последовало лишь в 1810 г.
Подобно фтору, основная масса хлора поступила на земную поверхность из горячих недр Земли. Даже в настоящее время с вулканическими газами ежегодно выделяются миллионы тонн и НСl и НF. Еще гораздо более значительным было такое выделение в минувшие эпохи.
Первичная форма нахождения хлора на земной поверхности отвечает его чрезвычайному распылению. В результате работы воды, на протяжении многих миллионов лет разрушавшей горные породы и вымывавшей из них все растворимые составные части, соединения хлора скапливались в морях. Усыхания последних привело к образованию во многих местах земного шара мощных залежей NаС1, который и служит исходным сырьем для получения соединений хлора.
Будучи наиболее практически важным из всех галоидов, хлор в громадных количествах используется для беления тканей и бумажной массы, обеззараживания питьевой воды (примерно 1,5 г на 1 м3) и в других отраслях техники. Ежегодное мировое потребление хлора исчисляется миллионами тонн.
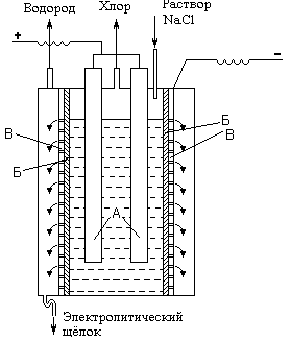
Рис.. Принципиальная схема электролизёра для получения хлора.
Основным промышленным методом получения хлора является электролиз концентрированного раствора NаС1. Принципиальная схема электролизера показана на рис. VII-5 (А - аноды, Б - диафрагма, В - катод). При электролизе на аноде выделяется хлор (2С1-- 2е- = С12), а в при катодном пространстве выделяется водород (2Н+ + 2е- = Н2) образуется NаОН.
При практическом осуществлении электролиза раствора NaCl расход электроэнергии на получение 1 т хлора составляет около 2700 кВт·ч. Полученный хлор под давлением сгущается в желтую жидкость уже при обычных температурах. Хранят и перевозят его в стальных баллонах, где он заключен под давлением около 6 атм. Баллоны эти должны иметь окраску защитного цвета с зеленой поперечной полосой в верхней части.
Для лабораторного получения хлора обычно пользуются действием MnO2 или КМnO4 на соляную кислоту:
МnО2 + 4 НСl = МnСl2 + Сl2 + 2 Н2О
2 КМnO4 + 16 НCl = 2 КCl + 2 МnСl2 + 5 Сl2 + 8 Н2О
Вторая реакция протекает значительно энергичнее первой (требующей подогревания).
Свободный хлор представляет собой желто-зеленый газ, состоящий из двухатомных молекул. Под обычным давлением он сжижается при -34 °С и затвердевает при -101 °С. Один объем воды растворяет около двух объемов хлора. Образующийся желтоватый раствор часто называют «хлорной водой».
Критическая температура хлора равна 144 °С, критическое давление 76 атм. При температуре кипения жидкий хлор имеет плотность 1,6 г/см3, а теплота его испарения составляет 20,5 кДж/моль. Твердый хлор имеет плотность 2,0 г/см3 и теплоту плавления 6,3 кДж/моль. Кристаллы его образованы отдельными молекулами С12 (кратчайшее расстояние между которыми равно 334 пм).
Связь Сl-Сl характеризуется ядерным расстоянием 198 пм. Термическая диссоциация молекулярного хлора по уравнению С12 + 242 кДж Ы 2 С1 становится заметной примерно с 1000 °С.
Атом хлора имеет в основном состоянии структуру внешнего электронного слоя 3s23р5 и одновалентен. Возбуждение его до ближайшего трехковалентного уровня 3s23р44s1 требует затраты
857 кДж/моль.
Энергия присоединения электрона к нейтральному атому хлора оценивается в 355 кДж/моль. Сродство к электрону хлора (аналогично и других галоидов) может быть вычислено при помощи рассмотрения реакций образования хлористых солей по отдельным стадиям. Например, для NаС1 имеем:
1) Nа (т) = Nа (г) — 109 кДж (теплота возгонки)
2) 1/2 С12 (г) = С1 (г) — 121 кДж (теплота диссоциации)
3) Na (г) = Nа+(г) + е- — 493 кДж (энергия ионизации)
4) С1(г) + е- = Сl-(г) + Х кДж (искомое сродство к электрону)
5) Nа+(г) + Сl-(г) = NаС1(т) +777 кДж (энергия кристаллической решетки)
в сумме: Nа(т) + 1/2 С12(г) = NаСl(т) + (Х+777-493-121-109) кДж
С другой стороны, непосредственно определенная на опыте теплота образования NаС1 из элементов равна: Nа(т) + 1/2 С12(г) = NаС1(т) + 410 кДж. Следовательно, по закону Гесса, Х + 777 - 493 - 121 - 109 = 410, откуда Х = 356 кДж.
Ион С1- — характеризуется эффективным радиусом 181 пм и энергией гидратации 351 кДж/моль. Для ковалентного радиуса хлора принимается половина ядерного расстояния молекулы С12, т. е. 99 пм.
Растворимость хлора в воде меняется с температурой следующим образом:
Температура, °С | 10 | 15 | 20 | 25 | 30 | 40 | 50 | 60 | |
Растворимость V на 1V H2O | 4,6 | 3,1 | 2,7 | 2,3 | 2,0 | 1,8 | 1,4 | 1,2 | 1,0 |
Описаны два кристаллогидрата хлора — С12·6Н2О и С12·8Н2О. В действительности они могут иметь переменный состав, так как являются клатратами.
Значительно хуже (примерно в 4 раза), чем в воде, растворяется хлор в насыщенном растворе NаС1, которым поэтому и удобно пользоваться при собирании хлора над жидкостью. Наиболее пригодным для работ с ним органическим растворителем является четыреххлористый углерод (СС14), один объем которого растворяет при обычных условиях около 50 объемов хлора.
Основным потребителями хлора являются органическая технология (получение хлорированных полупродуктов синтеза) и целлюлозно-бумажная промышленность (отбелка). Значительно меньше потребляется хлор в неорганической технологии, санитарной технике и других областях. Интересно недавно предложенное использование хлора для обработки металлов: под его действием с достаточно нагретой (инфракрасным излучением) поверхности все шероховатости удаляются в форме летучих хлоридов. Такой метод химической шлифовки особенно применим к изделиям сложного профиля. Было показано также, что струя хлора легко прорезает достаточно нагретые листы из жаростойких сплавов.
Хлор обладает резким запахом. Вдыхание его вызывает воспаление дыхательных путей. В качестве средства первой помощи при острых отравлениях хлором применяется вдыхание паров смеси спирта с эфиром. Полезно также вдыхание паров нашатырного спирта.
Предельно допустимой концентрацией свободного хлора в воздухе производственных помещений считается 0,001 мг/л. Пребывание в атмосфере, содержащей 0,01% хлора и выше, быстро ведет к тяжелому заболеванию. Признаком острого отравления является появление мучительного кашля. Пострадавшему необходимо прежде всего обеспечить полный покой; полезно также вдыхание кислорода.
По своей характерной химической функции хлор подобен фтору — он также является одновалентным неметаллом. Однако активность его меньше, чем у фтора. Поэтому последний способен вытеснять хлор из соединений.
Тем не менее химическая активность хлора очень велика —
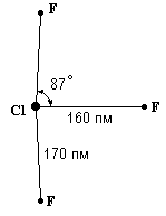
Рис.. Строение молекулы ClF3.
он соединяется почти со всеми металлами (иногда лишь в присутствии следов воды или при нагревании) и со всеми металлоидными элементами, кроме С, N и O. Важно отметить, что при полном отсутствии влаги хлор не действует на железо. Это и позволяет хранить его в стальных баллонах.Взаимодействие хлора с фтором при нагревании смеси сухих газов происходит лишь выше 270 °С. В этих условиях с выделением тепла (50 кДж/моль) образуется бесцветный хлорфторид — С1F (т. пл. -156, т. кип. -100 °C). Газообразный С1F обладает сильным своеобразным запахом (отличным от запахов хлора и фтора).
Взаимодействием хлорфторида с фторидами Сs, Rb и К под высоким давлением были получены бесцветные малостойкие соли типа МС1F2, содержащие в своем составе линейный анион С1F2-. При нагревании они экзотермически разлагаются около 250 °С.
Нагреванием ClF с избытком фтора может быть получен бледно-зеленоватый трехфтористый хлор (хлортрифторид) — СlF3 (т. пл. -76, т. кип. +12 °С). Соединение это также экзотермично (теплота образования из элементов 159 кДж/моль) и по запаху похоже на С1F. Молекула С1F3 полярна (m = 0,55) и имеет показанную на рис. плоскую структуру. Последняя производится от тригональной бипирамиды, у которой два направления треугольного основания закрываются свободными электронными парами атома хлора. Критическая температура С1F3 равна 154 °С, плотность в жидком состоянии 1,8 г/см3, теплота испарения 27,6 кДж/моль. Вблизи точки кипения пар трехфтористого хлора несколько ассоциирован по схеме: 2 С1F3Ы (С1F3)2 + 12,5 кДж. Для димера вероятна мостиковая структура (по типу F2С1F2С1F2).
Жидкий С1F3 смешивается с жидким НF в любых соотношениях, причем имеет место слабое взаимодействие по схеме: НF + С1F3Ы НС1F4 + 16,7 кДж. Образующийся ацидохлортетрафторид не выделен, но производящиеся от него соли типа МС1F4, (где М — Сs, Rb, К) известны. По-видимому, они могут быть получены не только прямым сочетанием МF и С1F3, но и фторированием соответствующих хлоридов (3000 атм, 300 °С).
Нагреванием смеси С1F3 с избытком фтора под высоким давлением может быть получен бесцветный хлорпентафторид — С1F5 (т. пл. -93, т. пл. -13 °С). Теплота его образования из элементов 251 кДж/моль. Молекула С1F5, имеет строение квадратной пирамиды из атомов фтора, вблизи основания которой располагается атом хлора. В отсутствие влаги этот газ при обычных условиях устойчив, а водой разлагается. Он является энергичным фторирующим агентом, но корродирует металлы слабее, чем С1F3.
Фториды хлора характеризуются исключительной реакционной способностью. Например, в парах С1F3 стеклянная вата самовоспламеняется. Почти столь же энергично взаимодействуют с ним и такие сами по себе чрезвычайно устойчивые вещества, как MgО, СаО, А12O3 и т. п. Так как С1F3 сжижается при обычных температурах уже под небольшим давлением и легко отщепляет фтор, его удобно использовать для транспортировки фтора. Помимо различных реакций фторирования, отмечалась возможность применения этого вещества как окислителя реактивных топлив и зажигательного средства в военной технике. По трифториду хлора имеется обзорная статья.
Взаимодействие хлора с водородом по реакции:
Н2 + С12 = 2 НС1 + 184 кДж
при обычных условиях протекает крайне медленно, но нагревание смеси газов или ее сильное освещение (прямым солнечным светом, горящим магнием и т. д.) сопровождается взрывом.
Детальное изучение этой реакции позволило выяснить сущность ее отдельных стадий. Прежде всего за счет энергии (hn) ультрафиолетовых лучей (или нагревания) молекула хлора диссоциирует на атомы, которые затем реагируют с молекулами водорода, образуя НСl и атом водорода. Последний в свою очередь реагирует с молекулой хлора, образуя НС1 и атом хлора, и т. д.:
1) С12 + hn = С1 + С1 (первоначальное возбуждение)
2)… С1 + Н2 = НС1 + Н
3)… Н+ С12 = НС1 + С1 и т. д.
Таким образом, получается как бы цепь последовательных реакций, причем за счет каждой первоначально возбужденной молекулы Сl2 образуется в среднем 100 тыс. молекул НС1. Реакции подобного типа называются цепными. Они играют важную роль при протекании многих химических процессов.
Фотохимическая диссоциация молекулы хлора на атомы вызывается светом с длиной волны 550 нм. Обеим стадиям цепной реакции образования хлористого водорода соответствуют следующие термохимические уравнения:
С1 + Н2 + 1 кДж = НС1 + Н и Н + С12 = НС1 + С1 + 188 кДж. Энергия активации первой из этих реакций составляет 25, а второй 8 кДж/моль. Малыми значениями этих энергий и обусловлено быстрое развитие цепи.
Очевидно, что цепь могла бы оборваться, если бы протекала реакция: Н + С1 = НС1. Такая возможность не исключена, однако вероятность осуществления этой реакции очень мала, так как концентрация атомов ничтожна по сравнению с концентрацией молекул и поэтому несравненно больше шансов имеет столкновение каждого из атомов с молекулой другого элемента, чем обоих атомов друг с другом. С другой стороны, произведенные на основе экспериментальных данных расчеты показывают, что даже при столкновении обоих атомов соединение между ними происходит далеко не всегда,
Рис 1-2 888888888
наоборот, процент успешных встреч очень мал. По этим же причинам цепи редко обрываются в результате реакций: С1+ Сl = С12 и Н + Н = Н2. Так, последняя из них осуществляется в газовой фазе лишь при одном столкновении из каждого миллиона.
“Огромное большинство реакций при ближайшем рассмотрении являются цепными реакциями” (Н. Н. Семенов). Это нередко вызывает отклонение их действительной молекулярности от отвечающей простейшему суммарному уравнению. В частности наблюдаемая на опыте бимолекулярность реакции образования волы из элементов обусловлена именно ее цепным характером: начало цепи дает (с энергией активации 188 кДж/моль) реакция Н2 + О2 = 2 ОН, после чего цепь разветвляется по схемам: ОН + Н2 = Н2О + Н, Н + О2 = ОН + О, О + Н2 = ОН + Н и т. д. Как видно из этих схем, число активных участников реакции (ОН, Н, О) последовательно возрастает, вследствие чего процесс протекает с самоускорением. Это и характерно для разветвленных цепных реакций, в отличие от неразветвленных, примером которых может служить синтез хлористого водорода.
Большие количества НС1 получают в технике как побочный продукт хлорирования органических соединений по схеме
RН + Cl2 = RС1 + НС1
где R — органический радикал. Однако для получения чистой соляной кислоты основное значение имеет прямой синтез. Исходным сырьем служат при этом хлор и водород, одновременно выделяющийся при электролизе раствора NаС1. Спокойное протекание процесса обеспечивается смешиванием обоих газов лишь в момент взаимодействия.
Еще один метод промышленного получения НС1 основан на взаимодействии NаС1 и концентрированной Н2SO4 по реакциям
NаС1 + Н2SO4 = NаНSO4 + НС1
NаС1 + NаНSO4 = Nа2SO4 + НС1
Первая из них протекает в значительной степени уже при обычных условиях и практически нацело — при слабом нагревании; вторая осуществляется лишь при более высоких температурах. Для проведения процесса служат специальные механизированные печи большой производительности.
Максимальная температура водородно-хлорного пламени составляет около 2200 °С. Для технического синтеза НС1 служит установка, схематически показанная на рис. Ч11 —8. После первоначального поджигания смесь хлора с водородом продолжает гореть спокойным пламенем, образуя хлористый водород. Последний проходит затем сквозь две поглотительные башни с водой, в которых и образуется соляная кислота. Используемый в системе принцип противотока, т. е. противоположных направлений движения газа и жидкости, обеспечивает полноту поглощения НС1 и позволяет проводить весь процесс непрерывно.
Основной частью показанной на рис. Ч11—9 механизированной печи для получения НСl является муфель А, со всех сторон обогреваемый горячими газами, идущими из топки Б. Внутри муфеля медленно вращается мешалка, гребенки которой устроены таким образом, что реагирующая масса передвигается ими от центра муфеля (куда подаются исходные вещества) к его краям. Выделяющийся хлористый водород после его обеспыливания и охлаждения улавливается водой, а образующийся Nа2SO4 сбрасывается в бункер Г (откуда грузится на вагонетки). Печь работает непрерывно и перерабатывает за сутки несколько тонн NаС1.
С теоретической стороны интересен метод получения хлористого водорода путем пропускания смеси хлора с водяным паром сквозь слой раскаленного угля. Реакция в этих условиях идет по уравнению:
2 Сl2 + 2 Н2О + С = СO2 + 4 HCl + 280 кДж
Так как она сильно экзотермична, уголь поддерживается в раскаленном состоянии за счет ее тепла. Практически этот метод не применяется (так как получающийся влажный хлористый водород сильно разъедает детали установки).
Хлористый водород (гидрохлорид) представляет собой бесцветный газ. В отсутствие влаги он при обычных температурах не действует на большинство металлов и их оксиды. Газообразный кислород окисляет его только при нагревании.
Молекула НСl характеризуется ядерным расстоянием d(HCl) = 128 пм, энергией связи 431 кДж. Хлористый водород плавится при -114 °С и кипит при -85 °С. Распад НС1 на элементы становится заметным примерно при 1500 °С.
Под давлением около 70 атм хлористый водород сжижается уже при обычных температурах и, подобно хлору, может транспортироваться к местам потребления а стальных баллонах. Жидкий хлористый водород обладает малой диэлектрической проницаемостью (4,6 при обычных температурах) и является плохим растворителем подавляющего большинства неорганических соединений. Растворимы в нем, например, хлориды олова и фосфора. Интересно, что РF3 растворим и жидком НС1, но не взаимодействует с ним, тогда как АsF3 и SbF3 испытывают полный сольволиз по схеме
ЭF3 + 3 НС1 = 3 НF + ЭС13
С темно-красным окрашиванием растворяется иод. Жидкий НС1 смешивается с жидкими СО2 и Н2S.
Предельно допустимой концентрацией хлористого водорода в воздухе производственных помещений считается 0,005 мг/л. Наличие уже 0,05 мг/л быстро вызывает раздражение в носу и гортани, колотье в груди, хрипоту и ощущение удушья. При хроническом отравлении малыми концентрациями НС1 особенно страдают зубы, эмаль которых подвергается быстрому разрушению.
Реакция в газовой фазе по уравнению
О2 + 4 НС1 = 2 Н2О + 2 С12 + 117 кДж
обратима. Ниже 600 °С равновесие ее смещено вправо, выше 600 °С — влево. На этой реакции был основан часто применявшийся ранее метод технического получения хлора: пропусканием смеси НС1 с воздухом над нагретым до 450 °С катализатором (пропитанный раствором СuС12 асбест) удавалось получать хлор с выходом около 70 % от теоретического. В связи с характерной для последнего времени дефицитностью хлора подобный метод может вновь приобрести промышленное значение.
На воздухе хлористый водород дымит вследствие образования с парами воды капелек тумана. Растворимость его весьма велика: при обычных условиях 1 объем воды способен поглотить около 450 объемов хлористого водорода.
Раствор НCl в воде называется хлористоводородной (иначе соляной) кислотой. Она относится к числу наиболее сильных кислот. Реактивная соляная кислота обычно имеет плотность 1,19 г/см3 и содержит около 37 % хлористого водорода. Состав ее близок к формуле HCl·3,5H2O.
Растворимость НС1 в воде меняется с температурой следующим образом:
Температура, °С | 10 | 15 | 20 | 25 | 30 | 40 | 50 | 60 | |
Растворимость в V на 1 V Н2О | 507 | 474 | 459 | 442 | 426 | 412 | 386 | 362 | 339 |
Растворение сопровождается выделением тепла (до 75 кДж/моль НСl). Давление хлористого водорода над крепкой соляной кислотой при 20 °С приводится ниже:
| Концентрация НСl, % | 24 | 26 | 28 | 30 | 32 | 34 | 36 | 38 |
| P, мм рт. ст. | 1,0 | 2,2 | 4,9 | 10,6 | 23,5 | 50,5 | 105 | 210 |
При смешивании концентрированной НС1 со снегом происходит резкое понижение температуры. Содержащий 25 вес.% НС1 водный раствор замерзает лишь при — 86 °С.
Органические жидкости поглощают хлористый водород гораздо хуже воды. Например, при обычных условиях эфир растворяет НС1 примерно в 3,5 раза, а бензол — в 50 раз меньше, чем вода.
Хлористый водород образует с водой азеотропную смесь, которая кипит под обычным давлением при 109 °С и содержит 20,2% НС1.
Охлаждением концентрированных водных растворов хлористого водорода могут быть выделены кристаллогидраты НСl с 6, 3, 2 и 1 молекулами Н2О, плавящиеся с разложением соответственно при -70, -25, -18, -15 °С. Последний из них по структуре является хлоридом оксония (Н3О+С1-), в кристаллогидрате НСl·2Н2О четко выявляются катионы Н5О2+ с очень короткой водородной связью [d(OO) = 241 пм] между двумя молекулами воды, а структура тригидрата соответствует формуле Н5О2+Сl-·Н2О. С жидким хлором хлористый водород дает молекулярные соединения состава С12·2НС1 и С12·НС1, плавящиеся соответственно при -121 и -115 °С.
Техническая соляная кислота выпускается крепостью не менее 31% НС1 (синтетическая) или 27 % HСl (из NаСl). Приблизительное процентное содержание НС1 в водном растворе легко найти, умножив на 2 число дробных долей его плотности. Например, при плотности 1,19 г/см3 процентное содержание получается равным 19·2 = 38 %. Следовательно, и обратно, зная процентное содержание НС1 в соляной кислоте той или иной крепости, можно приближенно оценить ее плотность. Путем приготовления 1,184 н. раствора НС1 удобно создавать среду с рН = 0 (при 25 °С). Как видно из приводимых ниже приблизительных данных, в крепких водных растворах (с моляльностью больше двух) коэффициент активности (f) хлористого водорода значительно превышает единицу:
m | 1 | 2 | 4 | 6 | 8 | 10 | 12 | 14 |
| f | 0,8 | 1 | 2 | 3 | 6 | 10 | 17 | 27 |
Соляная кислота очень сильно разъедает многие металлы. Транспортируют ее в стеклянных бутылях или гуммированных (т. е. покрытых слоем резины) металлических емкостях. Гуммирование может быть заменено введением в кислоту специальных добавок — ингибиторов.
Соляная кислота содержится в желудочном соке (около 0,3 %) и играет важную роль, так как способствует перевариванию пищи и убивает различные болезнетворные бактерии (холеры, тифа и др.). Если последние попадают в желудок вместе с большим количеством воды, то вследствие разбавления раствора НСl они выживают и вызывают заболевание организма. Поэтому во время эпидемий особенно опасна сырая вода. При повышении концентрации НС1 в желудке ощущается изжога, которую устраняют, принимая внутрь небольшое количество NаНСО3 или МgО. Наоборот, при недостаточной кислотности желудочного сока соляная кислота прописывается для приема внутрь (по 5-15 капель 8,3 %-ной НСl на 1/2 стакана воды до или во время еды).
Подобно другим сильным кислотам, НС1 энергично взаимодействует со многими металлами, оксидами металлов и т. д. Соли ее называются хлористыми или хлоридами. Большинство их хорошо растворимо в воде. Из производных наиболее обычных металлов труднорастворимы хлориды серебра и свинца. Ежегодное мировое потребление соляной кислоты исчисляется миллионами тонн. Широкое практическое применение находят также многие ее соли.
Длительное взаимодействие безводных СsС1 и НС1 при -78 °С ведет к образованию нестойкого СsС1·НС1 (давление пара > 400 мм рт. ст. при 30 °С). Хлориды других элементарных катионов подобных соединений не образуют, но было получено аналогичное производное катиона [N(СН3)4]+ и показано, что ион НСl2- аналогичен иону HF2-. В отличие от НF, образование такого иона [d(СlСl) = 314 пм] для HCl не характерно.
Непосредственное взаимодействие хлора с кислородом не приводит к образованию кислородных соединений хлора. Они могут быть получены лишь косвенными методами. Для рассмотрения путей их образования целесообразно исходить из обратимой реакции между хлором и водой:
С12 + Н2О + 25 кДж Ы НС1 + НОС1
При обычных условиях в насыщенном растворе гидролизовано около трети всего растворенного хлора.
Взаимодействие хлора с перекисью водорода первоначально протекает по уравнению:
С12 + Н2О2 = 2 НОС1
Однако избытком Н2О2 хлорноватистая кислота восстанавливается:
НОС1 + Н2О2 = НС1 + Н2О + О2
Из образующихся при гидролизе хлора двух кислот — соляной и хлорноватистой (HOС1) — первая является очень сильной, а вторая — очень слабой (слабее угольной). Это резкое различие в силе обеих кислот можно использовать для их разделения.
Если в растворе взболтать порошок мела (СаСО3) и затем пропускать в него хлор, то образующаяся соляная кислота реагирует с мелом по уравнению:
CaCO3 + 2 НС1 = СаС12 + СО2 + Н2О
а хлорноватистая накапливается в растворе. Подвергая реакционную смесь перегонке получают в приемнике разбавленный раствор НОС1.
Будучи соединением малоустойчивым, НОС1 медленно разлагается даже в таком разбавленном растворе. Соли хлорноватистой кислоты называются гипохлоритами. И сама HС1O, и ее соли являются очень сильными окислителями.
Наиболее концентрированные растворы НОС1 образуются при взаимодействии жидкого С12О с охлажденной водой (обе жидкости ограниченно растворимы друг в друге). Для получения растворов крепостью до 5 М удобно обрабатывать хлором (без избытка) взвесь оксида ртути в четыреххлористом углероде. Образующаяся в растворе С12О извлекается затем холодной водой. Возможно также получение раствора хлорноватистой кислоты по реакции:
2 С12 + Вi2О3 + Н2О = 2 ВiОСlЇ + 2 НОС1
Молекула НОС1 имеет угловое строение с параметрами d(НО) = 97, d(ОС1) = 169 пм, РНОС1 = 103°.
Хлорноватистая кислота обладает характерным запахом. Ее разбавленные растворы почти бесцветны, а более крепкие имеют желтый цвет. Константа кислотной диссоциации НОС1 при обычных условиях равна 4·10-8. Диссоциация ее по основному типу (т. е. НОСl Ы НО’+ С1•) экспериментально не обнаружена. Однако имеются косвенные указания на ее возможность. Например, с органическими соединениями НОС1 способна реагировать по схемам (R — органический радикал)
RН + НОС1 = RОН + НС1 и RН + НОСl = Н2О + RС1
т. е. и как окислитель, и как хлорирующее вещество.
Принципиальная возможность амфотерной диссоциации НОСl вытекает из общетеоретических соображений. Однако в присутствии С1’ непосредственно обнаружить С1• нельзя (из-за реакции по схеме С1•+ С1’ = С12·аq).
Так как при переходе от НОН к НОС1 отрицательный характер кислорода ослабевает, относительная вероятность внедрения в него протона уменьшается. Поэтому выражаемое схемой
ОН3+ + НОС1 Ы Н2О + Н2ОС1+
(или, учитывая неопределенную гидратированность обоих ионов, Н•+ НОСl Ы Н2О + Сl•) равновесие должно быть сильно смещено влево, но по мере повышения концентрации Н• должно несколько смещаться вправо. Экспериментально доказать возможность основной диссоциации НОС1 можно было бы, вероятно, подвергнув электролизу свежеприготовленный раствор С12О в холодной 30 %-ной серной кислоте: возникающий за счет приведенного выше равновесия положительный ион хлора должен был бы перемещаться к катоду.
Практический метод получения гипохлоритов основан на использовании приводимой выше обратимой реакции взаимодействия хлора с водой. Поскольку оба вещества правой части равенства — НСl и НОCl — дают в растворе ионы Н•, а оба исходных продукта — С12 и Н2О — таких ионов не образуют (точнее, почти не образуют), равновесие можно сместить вправо, связывая ионы Н•.
Добиться этого проще всего добавлением к реакционной смеси какой-нибудь щелочи. Так как по мере своего образования ионы Н• будут связываться ионами ОН’ в недиссоциированные молекулы воды, равновесие практически нацело сместится вправо. Применяя, например, КОН, имеем
С12 + Н2О Ы НОС1 + НС1
НОС1 + НС1 + 2 КОН = КОС1 + КС1 + 2 Н2О
или в общем: С12 + 2 КОН = КОС1 + КС1 + Н2О
В результате взаимодействия хлора с раствором щелочи получается, следовательно, смесь солей хлорноватистой и соляной кислот. Этот процесс имеет большое техническое значение, так как образующийся раствор гипохлорита обладает сильными окислительными свойствами и широко применяется для беления тканей (хлопковых и льняных) и бумаги.
Ввиду слабости хлорноватистой кислоты под действием углекислого газа воздуха происходит частичное ее выделение из раствора гипохлорита:
NаОС1 + CO2 + Н2О Ы NаНСО3 + НОС1
Беление основано на окислении хлорноватистой кислотой различных загрязняющих ткань веществ. Так как наличие NаС1 отбелке не вредит, применяют непосредственно раствор, получающийся в результате реакции хлора со щелочью.
Раствор этот часто называют “жавелевой водой”. На текстильных и бумажных фабриках ее иногда получают электролизом раствора NаС1 без диафрагмы. При этом первоначально образуются NаОН и С12, которые, взаимодействуя друг с другом, и дают “жавелевую воду”. После беления ею необходимо очень тщательно промывать ткани, так как избыток NаОС1 постепенно разъедает их.
Кристаллический натрийгипохлорит может быть получен отгонкой воды из его раствора под уменьшенным давлением, Выделяется он в виде кристаллогидрата NаОСl·5Н2О (т. пл. 45 °С), который легко переходит в NаОС1·Н2О. Последняя соль малоустойчива, а при нагревании до 70 °С разлагается со взрывом. Значительно устойчивее LiОС1·Н2О, который при обычных условиях выдерживает длительное хранение.
Опыт показывает, что окислительная активность гипохлоритов максимальна при таких значениях рН (близких к 7), когда в растворе одновременно имеются соизмеримые концентрации и ионов ОС1-, и молекул НОС1. Вероятно, это связано с равновесием по схеме:
ОСl- + НОСl Ы ОС1Н + ОСl-
Хотя оно и должно быть сильно смещено влево, его существование все же обеспечивает постоянную возможность временного возникновения неустойчивых молекул изохлорноватистой кислоты, структура которых позволяет предполагать наличие у них повышенной тенденции к отщеплению активного атома кислорода.
При взаимодействии хлора с более дешёвой щелочью — Са(ОН)2 (“гашёной известью”) — образуется хлорная известь. Реакция может быть приближенно выражена уравнением
С12 + Са(ОН)2 = Сl-Са-ОCl + H2O
согласно которому хлорная известь является смешанной солью соляной и хлорноватистой кислот. Она представляет собой белый порошок, обладающий сильными окислительными свойствами, и используется главным образом для дезинфекции.
Формула Са(С1)ОС1 отражает основной состав хлорной (иначе — белильной) извести лишь схематично. Получаемый хлорированием Са(ОН)2 продукт представляет собой смесь различных двойных и тройных соединений, образованных молекулами Са(ОСl)2, Са(ОН)2, СаС12 и кристаллизационной воды.
На воздухе хлорная известь постепенно разлагается, в основном по схеме:
2 Са(С1)ОСl + СО2 = СаС12 + СаСО3 + С12O
При действии на нее соляной кислоты выделяется хлор:
Са(С1)ОС1 + 2 НС1 = СаС12 + Н2О + С12
Этим иногда пользуются для его получения — хлорную известь смешивают с гипсом и из образовавшейся массы формуют кубики, которыми заряжают аппарат для получения газов. Качество хлорной извести оценивают обычно количеством хлора, образующимся при действии на нее соляной кислоты. Хорошие продажные сорта приближенно отвечают составу 3Са(С1)ОСl·Ca(ОН)2·nН2О и содержат около 35 вес.% «активного» (т. е. выделяющегося при действии соляной кислоты) хлора.
Для получения более высокопроцентной хлорной извести, состоящей главным образом из Са(ОС1)2, хлорированию подвергают не сухой Са(ОН)2, а взвесь его в небольшом количестве воды. При 30 °С реакция идет в основном по уравнению
2 Са(ОН)2 + 2 С12 = Са(ОС1)2 + СаС12 + 2 Н2О
причем большая часть образующегося Са(ОС1)2 выделяется в виде мелкокристаллического осадка. Получаемый после отфильтровывания и высушивания технический продукт содержит 45-70 % активного хлора. При взаимодействии с водой он растворяется почти полностью, тогда как обычная хлорная известь дает объемистый осадок Са(ОН)2.
Свободная хлорноватистая кислота испытывает в растворе три различных типа превращений, которые протекают независимо друг от друга. и поэтому называются параллельными реакциями:
1) НОС1 = НС1 + О
2) 2 НОС1 = Н2О + С12О
3) 3 НОС1 = 2 НС1 + НСlO3
Все эти процессы способны протекать одновременно, но их относительные скорости сильно зависят от имеющихся условий. Изменяя последние, можно добиться того, что превращение пойдет практически нацело по какому-нибудь одному направлению.
Под действием прямого солнечного света разложение хлорноватистой кислоты идет по первому из них. Так же протекает оно в присутствии веществ, способных легко присоединять кислород, и некоторых катализаторов (например, солей кобальта).
При нагревании крепкого раствора хлорной извести в присутствии солей кобальта распад ее идет по уравнению:
2 Са(С1)ОСl = 2 СаС12 + O2 + 92 кДж
Реакцией этой иногда пользуются для лабораторного получения кислорода.
При распаде по второму типу получается газообразный продукт — оксид хлора (С12О). Эта реакция идет в присутствии водоотнимающих веществ (например, СаС12). Оксид хлора представляет собой взрывчатый желто-бурый газ с запахом, похожим на запах хлора. При действии С12О на воду образуется НОС1, т. е. окись хлора является ангидридом хлорноватистой кислоты.
Молекула С12О полярна (m = 0,78) и характеризуется треугольной структурой [d(СlO) = -170 пм, Рa = 111°. Энергия связи О-С1 оценивается в 205 кДж/моль. Оксид хлора (дихлормоноксид) легко сгущается в красно-коричневую жидкость (т. пл. -121, т. кип. +2 °С), которая может длительно сохраняться при -78 °С, но более или менее быстро разлагается при обычных условиях (в основном по схеме 4 С12О = 2 С1О2 + 3 С12). Получать его удобно, действуя при охлаждении хлором на свежеосажденный сухую оксид ртути. Реакция идет по уравнению:
2 НgО + 2 Cl2 = С1НgОНgС1 + С12O + 79 кДж
Взрыв жидкого оксида хлора иногда происходит уже при переливании ее из одного сосуда в другой, а газообразной — при нагревании или соприкосновении со многими способными окисляться веществами. Он протекает по уравнению
2 С12О = 2 С12 + О2 + 150 кДж
Энергия активации этой реакции составляет 105 кДж/моль.
Оксид хлора хорошо растворим в СС14. Еще лучше он растворяется в воде за счет взаимодействия по реакции
Сl2O + Н2О Ы 2 НОС1
равновесие которой сильно смещено вправо (К = [С12О]/[НОС1]2 = 1·10-3 при 0 °С). Охлаждением крепких водных растворов С12О может быть получен кристаллогидрат хлорноватистой кислоты состава НОСl·2H2O (т. пл. -36 °С).
Распад НОС1 по третьему типу особенно легко идет при нагревании. Поэтому действие хлора на горячий раствор щелочи выражается суммарным уравнением
3 С12 + 6 КОН = КС1О3 + 5 КС1 + 3 Н2О
Продуктами реакции являются КС1 и калийная соль хлорноватой кислоты (НС1О3). Так как соль эта малорастворима в холодной воде, при охлаждении раствора она осаждается.
Свободная НС1О3 может существовать только в растворе. Она является сильной кислотой (диссоциированной приблизительно так же, как НС1 и НNО3) и энергичным окислителем. Соответствующий ей ангидрид неизвестен.
В противоположность свободной НС1О3, для ее солей (хлоратов) окислительные свойства в растворах не характерны. Большинство из них бесцветно (как и сама НС1О3) и хорошо растворимо в воде. Все они сильно ядовиты.
Переход гипохлорита в хлорат осуществляется, вероятно, с участием изохлорноватистой кислоты по схемам:
НСlO + СlO- = НСl + СlO2-и НСlO + СlO2 = НСl + СlO3-
Анион СlO3- имеет структуру треугольной пирамиды с хлором в вершине [d(ClO) = 145 пм, РОСlO = 106°].
Из солей хлорноватой кислоты практически наиболее важен КС1О3 (т. пл. 368 °С), который может быть получен электролизом горячего раствора КС1. Хлорат калия применяется в спичечном производстве, при изготовлении сигнальных ракет и т. д. Легкорастворимый в воде NаС1O3 (т. пл. 262 °С) является прекрасным средством для уничтожения сорных трав (на железнодорожном полотне и т. д.).
Энергия активации термического разложения чистого КС1О3 равна 226 кДж/моль (следует учитывать, что процесс этот может протекать со взрывом). Расплавленный КСlO3 энергично поддерживает горение. Смеси его с легко окисляющимися веществами (серой, фосфором, сахаром и др.) взрываются от удара.
Раствор хлорноватой кислоты обычно получают действием серной кислоты на Ba(ClO3)2 ( т. пл. 414 °С). Отфильтровав осадок ВаSO4, можно путем упаривания при низких температурах (в вакууме) сконцентрировать раствор примерно до 40 % содержания НС1О3. Получается густая бесцветная жидкость приблизительного состава НС1О3·7Н2О, при нагревании выше 40 °С разлагающаяся. Такой раствор характеризуется столь сильно выраженными окислительными свойствами, что при соприкосновении с ним бумага, вата и т. п. воспламеняются. Более разбавленные растворы НС1О3 в обычных условиях довольно устойчивы. При сильном охлаждении они становятся густыми и вязкими, но не закристаллизовываются.
При длительном совместном нагревании фторидов и хлоритов некоторых двухвалентных металлов в присутствии уксусной кислоты происходит взаимодействие по схеме
МF2 + M(С1O3)2 = 2 МС1O3F
с образованием соответствующей соли фторохлорноватой кислоты (Н2С1О3F). Таким путем синтезировались хорошо растворимые фторхлораты ряда лвухвалентных металлов (например, Сu(ClО3F·5H2O). Под действием на их растворы иона Са — осадок СаF2, начинает медленно выделяться лишь при кипячении, т. е. ион С1О3F” оказывается довольно устойчивым по отношению к гидролизу. Были получены также некоторые другие производные фторхлорноватой кислоты.
Осторожным восстановлением хлоратов может быть получен диоксид хлора (С1О2). Он представляет собой взрывчатый желтый газ, обладающий сильно выраженными окислительными свойствами.
В лабораторных условиях СlO2 удобно получать по реакции 2 КСlO3 + H2С2О4 = К2СО3 + СО2 + Н2О + 2 СlO2
нагреванием до 60 °С увлажненной смеси КСlO3 и щавелевой кислоты (Н2С2O4). Другим удобным методом лабораторного получения СlO2 является проводимая при 90 °С с тщательно осушенным хлором реакция по уравнению:
Сl2 + 2 АgСlO3 = 2 АgСl + 2 СlO2 + O2
При охлаждении ниже +10 °С диоксид хлора сгущается в красно-коричневую жидкость и может быть таким путем отделен от углекислого газа или кислорода.
Молекула С1О2 полярна (m = 1,78) и характеризуется треугольной структурой [d(СlO) = 147 пм, Рa = 118°]. Энергия связи С1-О равна 251 кДж/моль.
В твердом состоянии диоксид хлора (хлордиоксид) представляет собой желтовато-красные кристаллы (т. пл. -59 °С). Плотность ее пара отвечает простой формуле, но для раствора в СС14 установлено наличие частичной димеризации по схеме 2 С1O2Ы С12O4 (константа равновесия равна 0,18 при 25 °С). Запах ClO2 одновременно похож на запах хлора и азотной кислоты. Он начинает ощущаться при 0,002 %-ном содержании С1О2 в воздухе. В темноте чистый диоксид хлора устойчив по на свету или при наличии даже следов хлоридов постепенно разлагается. Будучи эндотермичным (теплота образования — 105 кДж/моль) и малоустойчивым соединением, С1О2 может взрываться при нагревании или соприкосновении со способными окисляться веществами.
Диоксид хлора хорошо растворим в воде (20: 1 по объему при 4 °С) с желто-оранжевой окраской жидкости. Разбавленные растворы (до 8 г/л) в темноте устойчивы но на свету медленно разлагаются (с образованием НСlO3 и НС1). Известен кристаллогидрат С1О2·6Н2О.
Используется С1О2 главным образом для отбелки или стерилизации различных материалов (бумажной массы, муки и др.). Установлено, что с его помощью можно производить обесфеноливание сточных вод химических заводов.
В связи с быстрым ростом потребления С1О2 для технических целей, был предложен ряд методов его промышленного получения. Примером может служить метод, основанный на экзотермической реакции
2 NаClO3 + SO2 + Н2SO4 = 2 NаНSO4 + 2 ClО2
проводимой с приблизительно 4 М серной кислотой (содержащей значительную примесь хлорид-иона).
Исходя из С1О2 довольно сложным путем было получено устойчивое -78 °С, но начинающее разлагаться уже при -45 °С темно-коричневое твердое вещество, состав которого отвечает формуле С12О3. Является ли оно действительно оксидом трехвалентного хлора (или представляет собой смесь других его оксидов), пока не ясно.
При медленном пропускании тока фтора под поверхность охлажденной до -50 °С диоксида хлора происходит ее фторирование с образованием фторхлордиоксида (FClO2). Вещество это представляет собой бесцветный газ (т. пл. -115, т. кип. -6 °С), довольно устойчивый по отношению к нагреванию, но весьма гигроскопичный. Гидролиз его идет по схеме:
FСlO2 + Н2О = НF + НСlO3
Взаимодействие FСlO2 с НС1 (при -110 °С) протекает по уравнению:
2 FСlO2 + 2 НСl = 2 НF + Сl2 + 2 СlO2
т. е. СlСlO2 оказывается совершенно неустойчивым. Вместе с тем были получены некоторые солеобразные производные СlO2+, например СlO2SbF6 (т. пл. 235 °С).
Взаимодействие С1О2 с раствором КОН медленно протекает по уравнению
2 С1О2 + 2 КОН = КС1О3 + КС1О2 + Н2О
с образованием солей двух кислот — хлорноватой и хлористой. Сама хлористая кислота (НС1О2) малоустойчива. По силе и окислительной активности она промежуточна между НОС1 и НС1О3. Соли ее (хлориты) используются при отбелке тканей.
Хлористую кислоту (К = 1·10-2) можно получить по реакциям:
ВаО2 + 2 С1О2 = Ва(С1О2)2 + О2 и
Ва(С1О2)2 + Н2SO4 = ВаSO4Ї +2 НС1O2
Она известна только в разбавленных растворах, при хранении которых очень быстро разлагается, в основном, по схеме:
4 HСlO2 = 2 СlO2 + НСlO3 + НCl + Н2О
Ион СlO2, имеет треугольную структуру [d(СlO) = 155 пм, РОСlO = 111°]. Хлориты, как правило, бесцветны и хорошо растворимы в воде [за исключением желтых АgСlO2 (1,7 г/л) и Рb(СlO2)2 (0,35 г/л при 0 °С)]. В отличие от гипохлоритов, они характеризуются наличием сильно выраженных окислительных свойств только в кислой среде. С другой стороны, под действием КМnO4 хлориты способны окисляться до хлоратов. Имеются указания на возможность образования некоторых хлоритов при непосредственном взаимодействии соответствующего металла (например, Ni) с раствором СlO2. В твердом состоянии многие соли НСlO2 легко взрываются при нагревании или ударе.
Наиболее практически важным хлоритом является NаСlO2. Эту соль удобно получать по реакции:
2 СlO2 + РbО + 2 NаОН = РbО2Ї + 2 NаСlO2 + Н2О
Выше 100 °С разлагается в основном по схеме:
3 NаСlO2 = 2 NаСlO3 + NаС1
При нагревании КС1О3 плавится, а около 400 °С начинает разлагаться, причем распад может идти по двум основным направлениям:
1) 4 КС1О3 = 4 КС1 + 6 О2 + 180 кДж
2) 4 КС1О3 = КС1 + 3 КС1О4 + 171 кДж
Реакция протекает преимущественно по первому типу при наличии катализатора (МnО2 и т. п.), по второму — в его отсутствие. Образующийся при распаде по второму типу хлорат калия) очень малорастворим в воде и поэтому легко отделяется от хорошо растворимого хлористого калия.
Действием на калийперхлорат концентрированной серной кислоты может быть получена свободная хлорная кислота (НС1О4), представляющая собой бесцветную, сильно дымящую на воздухе жидкость:
КС1О4 + Н2SO4Ы КНSO4 + НС1O4
Так как под уменьшенным давлением НС1O4 перегоняется без разложения, ее легко выделить из реакционной смеси.
Безводная НСlO4 малоустойчива и иногда взрывается просто при хранении, но ее водные растворы вполне устойчивы. Как окислитель HClO4 гораздо менее активна, чем НС1O3, и в разбавленных растворах практически не обнаруживает окислительных свойств. Напротив, кислотные свойства выражены у нее исключительно резко: по-видимому, она является одной из самых сильных кислот.
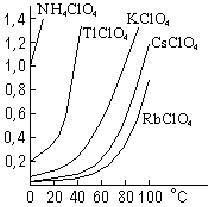
Рис.. Растворимость некоторых перхлоратов (моль/л H2O).
Соли НСlO4, за немногими исключениями (рис. ) легко растворимы в воде. Многие из них хорошо растворяются также в органических растворителях (спирте и т. п.). Подобно самой кислоте, большинство перхлоратов бесцветно.Калийперхлорат применяется для приготовления некоторых взрывчатых веществ. При 610 °С он плавится и одновременно начинает разлагаться, в основном по уравнению:
KСlO4 = КСl + 2 O2
Получают КСlO4 обычно электролизом раствора КСlO3. Реакция идет по схеме:
КСlO3 + Н2О = Н2 (катод) + КСlO4 (анод).
При перегонке разбавленных растворов НСlO4 сначала отгоняется вода, затем разбавленная кислота и, наконец, при 203 °С начинает перегоняться азеотропная смесь, содержащая 72 % HСlO4 (близкая к составу НСlO4·2Н2О и замерзающая лишь при -18 °C). Так как кипение последней сопровождается частичным разложением, перегонку HClO4 лучше проводить под уменьшенным давлением (при 20 мм рт. ст. азеотропная смесь перегоняется около 111 °С). Концентрированная (72%) кислота дымит на воздухе и весьма гигроскопична, но устойчива при хранении и не разлагается под действием света. Промышленностью обычно выпускается 30-70 %-ная НСlO4.
Молекула HСlO4 имеет форму пирамиды с тремя атомами кислорода в основании [d(СlO) = 141 пм], гидроксильной группой в вершине [d(С10) = 164 пм] и углом О-Сl=O, равным 106°. Безводная хлорная кислота (т. пл. — 101, т. кип. +16'С при 18 мм рт. ст.) представляет собой весьма подвижную жидкость, тогда как ее крепкие водные растворы имеют маслянистую консистенцию. Их охлаждением может быть получен плавящийся лишь при +50 °С кристаллогидрат НСlO4·Н2О, который следует рассматривать как перхлорат оксония — [Н3О]СlO4. Частичное образование последнего по схеме

Рис.. Электролитическая диссоциация HClO4.
3 НСlO4Ы [Н3О]СlO4 + Сl2O7 + 12,5 кДж(с константой равновесия К = 1·10-4) имеет место и в безводной хлорной кислоте. Именно этой реакцией (в силу последующего распада Сl2O7 по схеме 2 Сl2O7 = 4 СlO2 + 3 O2 + 117 кДж) обусловлена, вероятно, неустойчивость безводной хлорной кислоты. Очень сильные взрывы может вызвать ее соприкосновение со способными окисляться веществами. Хлорная кислота находит применение при анализах, в частности для выделения более летучих кислот из их солей.
В разбавленных водных растворах НСlO4 не восстанавливается такими сильными восстановителями, как НI, Н2S, SO2 и водород в момент выделения. Даже концентрированная кислота становится очень активным окислителем лишь при температуре кипения (когда она легко растворяет, в частности, специальные стали).
Хотя НСlO4 является одной из самых сильных из кислот, наличие недиссоциированных молекул в ее растворах установлено несколькими методами. Как видно из рис., заметным оно становится лишь в достаточно концентрированных растворах. Для константы равновесия НСlO4Ы Н+ + СlO4– получено значение К = 38. По другим данным, хлорная кислота ионизирована в растворах еще значительнее, чем то показано на рис. .
Входящий в состав перхлоратов анион СlO4- представляет собой тетраэдр с хлором в центре [d(СlO) = 144 пм].
Из безводных перхлоратов без разложения плавится только LiСlO4 (т. пл. 236 °С).
Вообще говоря, их термическое разложение может идти по двум схемам: с образованием хлорида металла и кислорода или оксида металла, хлора и кислорода. Для солей Сs, Rb, К характерен первый путь, для солей Nа, Li, Ва, Sr, Сa преимущественно он же, а для солей Мg и большинства других металлов основным становится второй путь распада.
Растворимость некоторых перхлоратов (г на 100 г растворителя при 25 °С) в воде, спирте и ацетоне сопоставлена ниже:
LiClO4 | NaClO4 | KClO4 | Mg(ClO4)2 | Ca(ClO4)2 | Ba(ClO4)2 | |
H2O | 60 | 210 | 2,1 | 100 | 189 | 198 |
C2H5OH | 152 | 15 | 0,01 | 24 | 166 | 125 |
(CH3)2CO | 137 | 52 | 0,16 | 43 | 150 | 125 |
Безводный перхлорат лития хорошо растворим и в эфире (с образованием 6 М раствора), тогда как кристаллогидрат LiСlO4·3Н2О растворим весьма мало. Следует отметить, что растворы перхлоратов в органических жидкостях, как правило, взрывоопасны.
Некоторые перхлораты (особенно NН4СlO4) используются в реактивной технике.
Взаимодействием 72 %-ной НСlO4 с фтором получен бесцветный фторперхлорат — FСlO4. Это малоустойчивое соединение (т. пл. -167, т. кип. -10 °С) обладает резким запахом и весьма реакционноспособно. И в газообразном, и в жидком состоянии оно может разлагаться со взрывом.
Длительным взаимодействием избытка СsСlO4 с ClSO3F при -45 °С был получен хлорперхлорат СlСlO4. Вещество это описывается как устойчивая лишь при низких температурах светло-желтая жидкость (т. пл. -117 °С). Наличие в молекуле хлорперхлората положительно поляризованного атома хлора устанавливается протекающими при -78 °С реакциями по схемам
НCl + СlOСlO3 = Сl2 + НСlO4 и
АgСl + СlOСlO3 = Сl2 + АgСlO4
Взрывоопасность СlСlO4 меньше, чем FСlO4.
Если фторперхлорат является продуктом замещения на фтор водорода хлорной кислоты, то в качестве продукта аналогичного замещения ее гидроксила можно рассматривать фторхлортриоксид (“перхлорилфторид”) — FСlO3. Последний образуется при действии фтора на сухой КСlO3 и представляет собой бесцветный газ (т. пл. -148, т. кип. -47 °С) с характерным сладковатым запахом. Удобнее получать его по схеме:
МСlO4 + НSО3F = МНSО4 + FСlO3
действием на перхлорат смеси хлорсульфоновой кислоты и SbF5 (которая играет роль катализатора). Теплота образования FСlO3, из элементов равна — 21 кДж/моль, а для энергий связей даются значения 251 (FСl) и 238 (СlO) кДж/моль. Молекула FСlO3 имеет структуру несколько искаженного тетраэдра с хлором около центра [d(СlO) = 140, d(FСl) = 161 А, РОС1O = 115°, РFСlO = 103°] и практически неполярна (m = 0,02).
Фторхлортриоксид термически устойчив до 400 °С, не гидролизуется даже горячей водой (и холодными щелочами), нерастворим в жидком фтористом водороде, умеренно токсичен и сам по себе невзрывчат (но способен давать взрывчатые смеси с некоторыми органическими веществами). Так как его критическая температура довольно высока (+95 °С), он может храниться и транспортироваться в сжиженном состоянии (при 25 °С давление пара составляет 12 атм). Окислительная активность FСlO3 в обычных условиях невелика, но быстро возрастает при нагревании. Поэтому реакции окисления им хорошо поддаются температурному регулированию. Вещество это представляет значительный интерес для реактивной техники. Существует также указание на то, что оно обладает наивысшим из всех газов значением диэлектрической проницаемости.
При слабом нагревании под уменьшенным давлением смеси безводной НС1О4 с фосфорным ангидридом (Р2О5) отгоняется бесцветная маслянистая жидкость, которая представляет собой хлорный ангидрид, образующийся по реакции
2 НСlO4 + Р2О5 = 2 НРО3 + Сl2O7
От сильного нагревания (и удара) Сl2O7 взрывается, однако он все же устойчивее, чем Сl2O и СlO2. При взаимодействии его с водой медленно образуется хлорная кислота.
Хлорный ангидрид (т. пл. -93, т. кип. 83 °С) является сильно эндотермичным соединением (теплота образования из элементов -251 кДж/моль). Строение его молекулы отвечает формуле О2-Сl-O-СlO3. Угол при кислородном атоме, соединяющем обе пирамиды СlO3 составляет 119° [при d(ОСl) = 171 пм], а угол O- Сl=О равен 115° [d(СlO) = 141 пм]. Молекула характеризуется отчетливо выраженной полярностью (m = 0,72). С такими веществами, как сера, фосфор, бумага, опилки и т. п., Сl2O7; при обычных температурах не реагирует, но соприкосновение его с иодом сопровождается взрывом. Хлорный ангидрид смешивается с четыреххлористым углеродом в любых соотношениях. При термическом разложении Сl2O7 первичным актом является разрыв одной из связей О-С1 (с образованием радикалов СlO3 и СlO4). Энергия этой связи оценивается в 201 кДж/моль.
Из двух радикалов, первично возникающих при термическом распаде хлорного ангидрида более или менее устойчивому существованию способен, по-видимому, лишь ClO3. Триоксид хлора (хлортриоксид) образуется при действии на СlO2 озона и представляет собой темно-красное масло (т. замерз. +3 °С). Жидкость примерно на 99 % состоит из удвоенных молекул (Сl2O6), тогда как в парообразном состоянии равновесие Сl2O6 + 8 кДж Ы 2 СlO3 очень сильно смещено вправо.
Хотя выше уже приводились названия кислородных кислот хлора и их солей, однако полезно сопоставить эти названия:
Кислота Формула Название солей
Хлорноватистая НОС1 гипохлориты
Хлористая НС1O2 хлориты
Хлорноватая НС1O3 хлораты
Хлорная НС1О4 перхлораты
Структурные формулы всех четырех кислот приводятся ниже:
Н-O-С1 Н-O-С1=O Н-O-С1=O O
ЅЅ ЅЅ
O H-O-Cl=O
ЅЅ
O
Как видно из этих формул, валентность хлора в рассматриваемых кислотах меняется по ряду: +1, +3, +5, +7.
Если сопоставить друг с другом кислородные кислоты хлора по важнейшим для них химическим свойствам — кислотности и окислительной активности, — получается следующая схема:
усиление кислотных свойств
——————————————®
НОС1 НС1О2 НС1О3 НС1О4
¬——————————————
увеличение окислительной активности
Кислотность изменяется, следовательно, противоположно окислительной активности. Последняя, в общем, тем больше, чем кислота менее устойчива. Действительно, хлорноватистая и хлористая кислоты более или менее устойчивы только в разбавленных растворах, концентрацию хлорноватой можно довести уже до 40 %, тогда как хлорная известна в безводном состоянии. Первые три кислоты в растворах постепенно разлагаются, а хлорная может сохраняться сколь угодно долго. Соответствующие соли обычно значительно устойчивее свободных кислот, но относительная их устойчивость примерно такова же.
Так как наиболее устойчивой из всех кислородных кислот хлора является НСlO4, можно было бы ожидать, что при взаимодействии хлора со щелочью должны сразу образовываться ее соли. Однако сперва получаются менее устойчивые соединения, которые затем лишь постепенно (быстрее — при нагревании) переходят в более устойчивые. На основе изучения ряда подобных случаев уже Гей-Люссак (1842 г.) наметил так называемое правило ступеней реакции: при химических процессах вначале обычно образуются не наиболее устойчивые вещества, а самые близкие по неустойчивости к исходной системе.
Во всех тех случаях, когда дальнейшие превращения относительно менее устойчивых продуктов реакции осуществляются очень быстро или, наоборот, очень медленно, мы практически их либо не замечаем, либо не считаем промежуточными продуктами. Поэтому выражаемое правилом ступеней реакции обобщение сразу бросается в глаза. Между тем при рассмотрении хода протекания химических процессов оно часто оказывается весьма полезным.
Подгруппа хрома.
По содержанию в земной коре хром (6·10-3 %), молибден (3·10-4 %) и вольфрам (6·10-4 %) относятся к довольно распространенным элементам. Встречаются они исключительно в виде соединений.
Хром был открыт в 1797 г., Mo — в 1778 г., W — в 1781 г.
Природный хром состоит из изотопов с массовыми числами 50 (4,3 %), 52 (83,8 %), 53 (9,5 %), 54 (2,4 %), молибден — из изотопов 92 (15,9 %), 94 (9,1 %), 95 (15,7 %), 96 (16,5 %), 97 (9,5 %), 98 (23,7 %), 100 (9,6 %), а вольфрам — из изотопов 180 (0,1 %), 182 (26,4 %), 183 (14,4 %), 184 (30,7 %), 186 (28,4 %).
Электронное строение атомов Cr (3d54s1) и Mo (4d55s1) соответствует их потенциальной шестивалентности уже в основном состоянии. Напротив, атом W (5d44s2) сам по себе четырёхвалентен, но возбуждение его шестивалентного состояния (5d56s1) требует затраты лишь 33,5 кДж/моль.
Наличие в почве следов молибдена, по-видимому, необходимо для нормального развития растительных организмов. Особенно это относится к растениям семейства бобовых. Вместе с тем установлено, что избыточное содержание молибдена в корме рогатого скота вызывает желудочные заболевания, а избыток его в продуктах питания человека способствует развитию подагры.
Основной рудой хрома является природный хромистый железняк (FеО·Сr2О3). Из молибденовых руд наиболее важен минерал молибденит (МоS2), из руд вольфрама — минералы вольфрамит (хFеWO4·уМnWO4) и шеелит (СаWO4).
Ежегодная мировая (без России) добыча хрома (в рудах) составляет около 2 млн. т. Молибдена и вольфрама добывается примерно по 50 тыс. т.
При получении элементов подгруппы хрома первой задачей является выделение их оксидов. Для этого обычно пользуются следующими схемами процессов. Хромистый железняк сплавляют с содой в присутствии кислорода воздуха:
4 (FеО·Сr2О3) + 8 Nа2СО3 + 7 O2 = 2 Fе2О3 + 8 Nа2СrO4 + 8 СО2,
после чего выделенный из сплава Nа2СrO4 переводят в Na2Сг2О7 по схеме:
2 Nа2СrO4 + Н2SО4 = Nа2SO4 + Nа2Сr2O7 + Н2О,
а последний восстанавливают до Сr2О3 углем:
Nа2Сr2О7 + 2 С = Сr2О3 + Nа2СО3 + СО.
Полученный из вольфрамита путем подобного же сплавления с содой по реакциям:
4 FеWO4 + 4 Nа2СО3 + O2 = 4 Nа2WO4 + 2 Fе2О3 + 4 СО2 и
6 МnWO4 + 6 Nа2СО3 + O2 = 6 Nа2WО4 + 2 Мn3O4 + 6 СО2
вольфрамат натрия разлагают соляной кислотой и выделившуюся Н2WO4 прокаливают до перехода ее в WO3. Молибденит переводят в MoО3, обжигом на воздухе:
2 МоS2 + 7 O2 = 4 SO2 + 2 МоО3
Для получения элементарного хрома удобно исходить из смеси его оксида (Сr2О3) c порошком алюминия. Начинающаяся при нагревании реакция идёт по уравнению
Сr2О3 + 2 Аl = Аl2O3 + 2 Сr + 539 кДж
Молибден и вольфрам могут быть получены восстановлением их оксидов при высоких температурах углем или водородом.
При алюмотермическом получении хрома к исходной Сr2О3 обычно добавляют немного СrО3, (чтобы процесс протекал энергичнее). В результате реакции образуются два слоя, из которых верхний содержит красный (от следов оксида хрома) оксид алюминия, а нижний — примерно 99,5 %-ный хром. Восстановление МоО3 и WO3 водородом до металлов легко идет выше 500 °С. Из руд Сr, Mo и W обычно выплавляют не чистые металлы, а их высокопроцентные сплавы с железом. Исходным материалом для приготовления феррохрома (не менее 60 % Сr) является непосредственно хромистый железняк. Молибденит предварительно переводят в МоО3, исходя из которого затем и готовят ферромолибден (не менее 55 % Mo). Для получения ферровольфрама (65–80 % W) могут служить бедные марганцем вольфрамиты.
В компактном виде элементы подгруппы хрома представляют собой серовато-белые блестящие металлы. Их важнейшие константы сопоставлены ниже:
| Cr | Mo | W | |
Плотность, г/см3 | 7,2 | 10,2 | 19,3 |
Температура плавления, °С | 1875 | 2615 | 3387 |
Температура кипения, °С | 2570 | 4830 | 5370 |
| Относительная электропроводность (Нg = 1) | 5 | 20 | 18 |
Очень чистые металлы хорошо поддаются механической обработке, но уже следы примесей сообщают им твёрдость и хрупкость. Технический хром чрезвычайно твёрд. Молибден и вольфрам значительно мягче. По отношению к воздуху и воде Сr, Mo и W при обычных условиях вполне устойчивы. Их основным потребителем является металлургическая промышленность, где эти металлы используются в производстве специальных сталей.
Теплоты плавления рассматриваемых элементов составляют 13,8 (Cr), 27,6 (Mo) и 35,1 (W) кДж/моль, теплоты испарения — 347 (Сr), 594 (Mo) и 798 (%) кДж/моль, теплоты атомизации (при 25 °С) — 397 (Cr), 660 (Mo) и 853 (W) кДж/моль. У хрома при 1840 °С отмечен переход из одной аллотропической формы в другую (теплота перехода 1,67 кДж/моль).
Очень чистый хром может быть получен, например, перегонкой электролитически осаждённого металла в высоком вакууме. Он пластичен, однако уже при хранении на воздухе поглощает следы газов (О2, N2, Н2) и теряет пластичность.
Введение Cr, Mo и W в состав сталей сильно увеличивает их твердость. Такие стали применяются главным образом при изготовлении ружейных и орудийных стволов, броневых плит, рессор и режущего инструмента. Обычно эти стали очень устойчивы также по отношению к различным химическим воздействиям. Примесь молибдена была обнаружена в старинных японских мечах, а вольфрама — в дамасских кинжалах. Уже небольшая присадка молибдена (порядка 0,25 %) сильно улучшает механические свойства чугуна.
Сталь с содержанием 15-18% W, 2-5 % Сu и 0,6-0,8 % С может быть сильно нагрета без потери твердости. При содержании более 10 % Сr сталь почти не ржавеет. Поэтому из неё делают, в частности, лопатки турбин и корпуса подводных лодок. Сплав 35 % Fе, 60 % Сr и 5% Mo отличается своей кислотоупорностью. Ещё в большей степени это относится к сплавам Mo с W, которые могут во многих случаях служить для замены платины. Сплав W с Аl (”партиниум”) применяется при изготовлении автомобильных и авиационных моторов. Сплавы на основе молибдена сохраняют механическую прочность при весьма высоких температурах (но нуждаются в защитном от окисления покрытии).
Помимо введения в специальные стали, хром используется для покрытия металлических изделий, поверхность которых должна оказывать большое сопротивление износу (калибры и т. п.). Подобное хромирование осуществляется электролитическим путем, причем толщина наносимых плёнок хрома, как правило, не превышает 0,005 мм. Металлический молибден применяется главным образом в электровакуумной промышленности. Из него обычно делают подвески для нитей накала электроламп. Попеременным опусканием и извлечением конца тонкой молибденовой или вольфрамовой проволоки в расплавленный нитрит натрия (NаNО2) можно получать тончайшие острия.
Так как вольфрам является наиболее тугоплавким из всех металлов, он особенно пригоден для изготовления нитей электроламп, некоторых типов выпрямителей переменного тока (так называемых кенотронов) и антикатодов мощных рентгеновских трубок. Громадное значение имеет вольфрам также для производства различных сверхтвёрдых сплавов, употребляемых в качестве наконечников резцов, сверл и т. д.
Рис. 1. Относительные характеристики ламп накаливания.
Лампы накаливания являются в настоящее время основным средством искусственного освещения. Для повышения коэффициента их полезного действия температура нити накала должна быть возможно более высокой (так как световая отдача раскалённого тела пропорциональна четвертой степени его абсолютной температуры). В современных электролампах нити накала работают при температурах около 2600 °С, что возможно лишь благодаря исключительной тугоплавкости и нелетучести вольфрама. Как видно из рис. 1, отклонения в ту или иную сторону от нормального для данной лампы напряжения (принятого за единицу) существенно сказываются и на её световой отдаче, и на сроке службы. Мировое производство электроламп исчисляется миллиардами штук ежегодно.При длительной работе обычной электролампы вольфрам с её нити постепенно испаряется и оседает тёмным слоем на стекле, а становящаяся все более тонкой нить накала наконец перегорает. Этот процесс “старения” можно сильно задержать введением в лампу следов иода: образующийся при сравнительно невысоких температурах летучий WI2 затем разлагается на накалённой нити, тем самым возвращая ей испарившийся металл. Подобные “иодные лампы” могут при очень малых размерах быть гораздо ярче обычных (за счёт повышения температуры накала), причем их близкий по спектральному составу к дневному световой поток постоянен в течение всего срока службы. Они работают в стационарном режиме уже через 1/2 сек после включения и передают тепло в окружающее пространство более чем на 80 % лучеиспусканием. Мощные установки такого типа с успехом используются для нагревательных целей, вообще же впервые реализованные в 1959 г. иодные лампы уже находят самые разнообразные области применения. Обычно их делают из кварцевого стекла и заполняют (под давлением в несколько атмосфер) ксеноном с примесью паров иода. Важно, чтобы все внутренние металлические детали были только вольфрамовыми.
Работа широко применяемого в практике кенотронного выпрямителя основана на способности сильно нагретых металлов испускать электроны. Простейший кенотрон (рис. 2) представляет собой вакуумированный стеклянный баллон, содержащий два электрода: один — в виде вольфрамовой спирали (А), другой — в виде пластинки (Б). Если такой прибор с накаленной (от отдельного источника тока) спиралью включить в цепь переменного тока, то при минусе на спирали электроны переходят на второй электрод, и во внешней цепи идет ток. Напротив, при плюсе на спирали внешняя цепь остается разомкнутой. Таким образом, направление тока все время сохраняется неизменным, т. е. переменный ток превращается в постоянный (точнее, пульсирующий постоянный). Основное преимущество кенотронов перед другими видами выпрямителей заключается в возможности выпрямлять при их помощи тока весьма высокого напряжения. Работа выхода электрона составляет для вольфрама 4,5 эВ, а для молибдена 4,3 эВ.
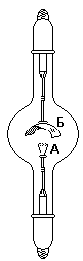
Рис. 2. Схема
кенотрона.
Сверхтвердые сплавы (“победит” и т. п.) содержат обычно 80-87 % W, 6–15 % Со и, 5-7 % С. Изготовляются они методом порошковой металлургии. Сущность этого метода заключается в накаливании до спекания спрессованной смеси порошкообразных исходных веществ (иногда с её последующей механической обработкой в горячем состоянии). Так как спекание осуществляется при гораздо более низких температурах, чем плавление данного вещества, метод порошковой металлургии часто используется и для изготовления (обычно под давлением) различных металлических изделий.
С помощью порошковой металлургии получают, в частности, весьма важные для современной техники материалы на основе сочетания огнеупорных веществ (как правило, оксидов) с металлами. Подобные материалы — керметы — характеризуются особой стойкостью при высоких температурах. Состав их может быть очень разнообразен. Например, был предложен кермет, состоящий из 83 % Сr2О3, 2 % WС и 15 % Ni, в котором никель играет роль связки между частицами двух других веществ. Важной группой керметов являются обладающие высокой термической стойкостью и хорошей прочностью композиции из хрома и оксида алюминия (например, 72 % Сr и 28 % Аl2O3).
В обычных условиях все три металла заметно взаимодействуют лишь с фтором, но при достаточном нагревании более или менее энергично соединяются и с другими типичными металлоидами. Общим для них является отсутствие химического взаимодействия с водородом.
Электролитически осажденный хром содержит много (порядка десятков объемов) растворенного водорода, который прочно удерживается металлом в обычных условиях, но может быть удалён нагреванием под вакуумом. При определённых условиях электролиза получается гидрид точного состава СrН, характеризующийся определённой кристаллической структурой [в которой водород гексакоординирован и d(CrН) = 191 пм]. Из газовой фазы хром начинает заметно поглощать водород лишь при высоких температурах. То же, но в значительно меньшей степени, относится к молибдену и вольфраму.
Вместе с тем для вольфрама существует гидрид состава WН6·2X. [где X — Р(CН3)2С2Н5], в известной мере аналогичный рениогидридам. Он был получен восстановлением WСl4·2X раствором NаВН4 в метиловом спирте и представляет собой белое кристаллическое вещество (т. пл. 112 °С с разл.), хорошо растворимое в органических растворителях. При действии на него разбавленной НС1 происходит выделение водорода с образованием исходного WСl4·2X.
В ряду напряжений хром располагается между Zn и Fе; между тем, на холоду внесенный в НС1 металл начинает растворяться не сразу. Обусловлено это наличием на его поверхности тончайшего (и поэтому незаметного), но очень плотного слоя химически малоактивного оксида (Сr2О3), препятствующего взаимодействию металла c кислотой. Оксид этот растворяется в НС1 при нагревании и может быть удалён также простым соскабливаем погружённой в жидкость поверхности. Однако на воздухе к хрому возвращается его пассивность. Таким образом, по существу хром на воздухе окисляется, но практически окисление незаметно, так как образовавшийся слой оксида предохраняет металл от дальнейшего разрушения. С образованием подобной защитной пленки связана и пассивность хрома по отношению к азотной кислоте и другим окислителям. Пассивирование под действием окислителей довольно характерно также для молибдена и вольфрама. При температуре красного каления Сr, Mo и W взаимодействуют с водяным паром, вытесняя водород.
При переходе в подгруппе сверху вниз (Сr®Mo®W) химическая активность металлов уменьшается. Особенно наглядно сказывается это на их отношении к кислотам. Хром растворим в разбавленных НСl и Н2SО4. На молибден они не действуют, но в горячей крепкой Н2SO4 металл этот растворяется. Вольфрам устойчив по отношению ко всем обычным кислотам и их смесям (кроме НF + НNО3). Перевод молибдена и вольфрама в растворимое соединение легче всего осуществляется путем сплавления с селитрой и содой по схеме
Э + 3 NаNО3 + Nа2СО3 = Na2ЭO4 + 3 NаNO2 + СO2
Для элементов подгруппы хрома известны соединения, отвечающие различным валентностям, вплоть до VI. Из всех них сколько-нибудь значительное применение находят только производные шестивалентных элементов и трёхвалентного хрома, причем важнее других хромовые препараты. Соединения низших степеней окисления Mo и W ещё сравнительно плохо изучены.
Суммарным переходам по схеме Э+6 + 6 е- = Э отвечают следующие окислительно-восстановительные потенциалы в кислой (первая цифра) и щелочной (вторая цифры) средах: +0,29 и -0,51 (Сr), 0,0 и -1,05 (Mo), -0,09 и -1,25 (W). Наиболее типичные для хрома изменения значности характеризуются приводимыми ниже потенциалами:
Значность… 0 +2 +3 +6
Кислая среда… -0,91 -0,41 +1,33
Щелочная среда… -1,4 -1,1 -0,13
Все производные шестивалентного хрома сильно ядовиты. При попадании на кожу или слизистые оболочки они вызывают местное раздражение (иногда с образованием язв), а при вдыхании в распылённом состоянии способствуют возникновению рака легких. Предельно допустимым их содержанием в воздухе производственных помещений считается 0,0001 мг/л.
Наиболее характерны для элементов подгруппы хрома те производные, в которых они шестивалентны. Из отвечающих этой валентности триоксидов (ЭО3) при прокаливании металлов на воздухе образуются лишь бесцветный МоО3 и светло-желтый WO3. Темно-красный СrО3 может быть получен только косвенным путем (исходя из более сложных соединений). Все эти триоксиды при обычных условиях — твердые вещества.
Будучи типичным кислотным ангидридом, СгО3 растворяется в воде с образованием характеризующейся средней силой хромовой кислоты — Н2СrO4. Хромовый ангидрид ядовит и является очень сильным окислителем. Уже выше 200 °С он начинает разлагаться по суммарной схеме:
4 СrО3 = 2 Сr2О3 + 3 O2
Напротив, МоО3 и WO3 около 1000 °С испаряются без разложения.
Теплота образования из элементов возрастает по ряду СrО3 (577) - МоО3 (752) - WO3 (840 кДж/моль). При нагревании триоксида молибдена (т. пл. 795, т. кип. 1155 °С) он желтеет, а триоксид вольфрама (т. пл. 1470 °С под давл.) становится оранжевым. Первый из этих оксидов начинает заметно возгоняться выше 650 второй — выше 850 °С. В парах оба они частично полимеризоваиы (с образованием преимущественно молекул Э3О9). И для МоО3, и для WO3 известны продукты присоединения фторидов щелочных металлов (кроме лития) типа ЭО3·МF. Описаны также представители типа WО3·3МF, где М — Сs, Rb, К.
В присутствии водяного пара летучесть МоО3 и WO3 заметно повышается. Обусловлено это, по-видимому, газофазными реакциями типа ЭО3 + Н2О Ы ЭО2(ОН)2 равновесия которых при высоких температурах смещаются вправо, а при более низких — влево. Вероятно, то же относится и к летучести твердого СгО3.
Характер промежуточных продуктов термического разложения СrО3 (т. пл. 196 °С) зависит от условий проведения этого процесса, Области их устойчивости приблизительно соответствуют схеме:
СrО3(220 °C) ® Сr3О8(280 °C) ® Сr2О5(370 °C) ® СrО2(450 °C) ® Сr2О3
Отмечалось существование окcидов и более сложного состава.
Растворимость МоО3 и WO3 в воде очень мала, но в щелочах они растворяются с образованием солей молибденовой и вольфрамовой кислот. Последние в свободном состоянии представляют собой почти нерастворимые порошки белого (Н2МоО4) или желтого (Н2WO4) цвета. При нагревании обе кислоты легко отщепляют воду и переходят в соответствующие триоксиды.
Рис. 3. Растворимость хроматов и бихроматов.
Растворимость МоО3 и WO3 в воде составляет соответственно около 0,4 и 0,02 г/л. Константы первой ступени кислотной и основной диссоциации Н2МоО4 имеют порядок соответственно 10-2 и 10-13. Вольфрамовая кислота известна в двух формах — белой и желтой. Первая осаждается при взаимодействии растворов вольфрамовокислых солей с разбавленными сильными кислотами на холоду, вторая — с более крепкими сильными кислотами при нагревании. Менее устойчивая белая форма постепенно переходит в желтую. Первая из них представляет собой, по-видимому, коллоидный осадок переменного состава (WO3·хН2О), вторая — определенное химическое соединение (H2WO4·Н2О). Ионы МоO42- и WO42- имеют структуру тетраэдров с атомом металла в центре [d(МоО) = 177, d(WO) = 179 пм].По ряду Сr-Mo-W сила кислот Н2ЭО4 уменьшается. Большинство их солей малорастворимо в воде. Из производных чаще встречающихся металлов хорошо растворимы: хроматы — лишь Nа+, К+, Mg2+ и Са2+, молибдаты и вольфраматы — только Nа+ и К+. Хромовокислые соли окрашены, как правило, в светло-желтый цвет иона СrO42-, молибденово- и вольфрамовокислые бесцветны.
Кроме кислот типа Н2ЭO4 для хрома и его аналогов существуют также отвечающие общей формуле Н2Э2О7 и по строению аналогичные пиросерной кислоте. Наибольшее значение из них имеет двухромовая кислота — Н2Сr2О7. Сама она известна только в растворе, но ее соли (двухромовокислые, или бихроматы), К2Сr2О7 (“хромпик”) и Nа2Сr2О7·2Н2О — особенно, являются наиболее обычными хромовыми препаратами и исходными продуктами для получения остальных соединений этого элемента.
Прокаливание безводного Nа2Сr2О7 (т. пл. 320 °С) выше 400 °C сопровождается его термическим разложением по уравнению:
4 Nа2Сr2О7 = 4 Nа2СrO4 + 2 Сr2О3 + 3 O2
Хромпик (т. пл. 398 °С) разлагается аналогично, но лишь при более высоких температурах. Хроматы натрия и калия плавятся соответственно при 790 и 963 °С, т. е. значительно выше бихроматов. Ион СrО42- имеет структуру тетраэдра с d(СrО) = 165, а ион Сг2О72- — структуру О3Сr-O-СrО3 с РСrОСr = 115° и d(ОСr) = 191 пм.
Для константы равновесия К = [Сг2О72-]/[НСгО4-] при 20 °C в зависимости от ионной силы раствора даются значения 100 (m = 1,0) или 80 (m= 0,5). Хромовая кислота (К1 = 2·10-2 и К2 = 3·10-7) значительно слабее двухромовой (К2 = 2·10-2). Последняя является простейшим представителем так называемых изополикислот общей формулы mН2О·nЭО3 (где n>m), известных в виде их солей. Помимо оранжево-красных бихроматов (m = 1, n = 2), получены темно-красные трихроматы (m = 1, n = 3) и коричнево-красные тетрахроматы (m = 1, n = 4). Из них К2Сr3O10 и К2Сr4O13 плавятся с разложением (переходя в К2Сr2О7) соответственно при 243 и 210 °С. Образование подобных и более сложных изополисолей особенно характерно для молибдена и вольфрама. По мере усложнения состава изополикислот сила их обычно возрастает.
Предельным типом изополикислот (n = ![]() ) является соответствующий свободный ангидрид. Из относящихся сюда соединений МоО3 и WO3 образуются непосредственно при прокаливании металлов на воздухе. Игольчатые кристаллы СrO3 выделяются при стоянии смеси 10 %-ного раствора К2Сг2О7 с концентрированной Н2SO4 (4: 1 по объему).
) является соответствующий свободный ангидрид. Из относящихся сюда соединений МоО3 и WO3 образуются непосредственно при прокаливании металлов на воздухе. Игольчатые кристаллы СrO3 выделяются при стоянии смеси 10 %-ного раствора К2Сг2О7 с концентрированной Н2SO4 (4: 1 по объему).
Взаимодействие по схеме:
4 СrО3 + 6 Н2SO4 + 3 С2H5ОН = 2 Сr2(SO4)3 + 3 CН3СООН + 9 Н2О
используется для обнаружения алкоголя в выдыхаемом воздухе. Индикатором его наличия служит позеленение реактива вследствие образования Сr2(SO4)3.
Подобно самому иону Сr2О72-, большинство бихроматов имеет красно-оранжевую окраску. Растворимость их в общем выше, чем соответствующих хроматов. Данные для солей натрия и калия приведены на рис. 2.
Растворы бихроматов показывают кислую реакцию, обусловленную тем, что ион Сr2O72- реагирует с водой по схеме:
Н2О + Сr2О72-Ы 2 НСrO7-Ы 2 Н+ + 2 СrO42-.
Как видно из уравнения, прибавление к раствору кислот (ионов Н+) должно смещать равновесие влево, а прибавление щелочей (ионов ОН-) — вправо. В соответствии с этим из бихроматов легко получить хроматы и наоборот, например, по реакциям:
К2Сr2О7 + 2 КОН = 2 К2СrO4 + Н2О
2 К2СrO4 + Н2SO4 = К2SO4 + К2Сr2О7 + Н2О
Соли хромовых кислот в кислой среде являются сильными окислителями (СrVI восстанавливается до СrIII). Например, ими уже на холоду окисляется НI, а при нагревании — НВг и даже НСI. Реакции идут по схеме
К2Сr2О7 + 14 НГ = 2 КГ + 2 СrГ3 + 3 Г2 + 7 Н2О
Обладающая очень сильным окислительным действием смесь равных объемов насыщенного на холоду раствора К2Сr2О7 и концентрированной Н2SO4 (“хромовая смесь”) применяется в лабораториях для мытья химической посуды.
Из отдельных проявлений высокой окислительной активности шестивалентного хрома следует отметить взаимодействие хромпика с крепкой соляной кислотой по уравнению:
К2Сr2О7 + 14 НС1 = 2 КС1 + 2 СгСl3 + 3 Сl2 + 7 Н2О
Реакция эта интересна тем, что она идёт только при нагревании и поэтому удобна для получения хлора в небольших количествах, так как при прекращении нагревания прекращается и выделение газа. С методической стороны интересна реакция по уравнению:
2 Н2СrO4 + 3 Н2SО3 = Сr2(SO4)3 + 5 Н2О
Здесь из двух кислот получается типичная соль.
Действием очень сильных восстановителей производные СrVI могут быть восстановлены в нейтральной и даже слабощелочной среде. Так, например, при нагревании идёт важная для аналитической химии реакция с сульфидом аммония:
2 К2СrO4 + 3 (NН4)2S + 8 Н2О = 2 Сr(ОН)3 + 3 S + 4 КOH + 6 NН4OН
В противоположность хрому, шестивалентные Mo и W даже в кислой среде могут быть восстановлены только сильными восстановителями. В частности, при действии водорода в момент выделения на растворы соответствующих солей последовательно образуются различно окрашенные соединения низших степеней окисления.
Для всех элементов подгруппы хрома характерно образование при взаимодействии с Н2О2 пероксидных соединений. Сам хром помимо синего пероксида СrО5 (образующегося в кислой среде по уравнению Н2СrO4 + 2 Н2О2 = 3 Н2О + СrО5) даёт соли надкислот Н2СrО6 и Н2СrО8. Результаты изучения химических и магнитных свойств рассматриваемых соединений говорят в пользу следующих структурных формул:
Таким образом, валентность хрома в обеих надкислотах оказывается различной.
Рис. 4. Строение иона CrO83–.
Соли первой из них обычно окрашены в синий, соли второй — в красный цвет. В зависимости от условий образуются те или другие. Так, осторожным добавлением 30 %-ной Н2О2 к охлажденному до 0 °С раствору К2Сr2О7 могут быть получены сине-фиолетовые кристаллы КНСrО6·Н2О, медленно разлагающиеся уже при обычной температуре. Коричнево-красные кристаллы К3СrО8 могут быть получены действием 30 %-ной Н2О2 на содержащий большой избыток КОН раствор К2СrO4. При обычных условиях они довольно устойчивы и быстро (со взрывом) разлагаются лишь выше 170 °С. Строение иона СrО83- показано на рис. 4.В водном растворе все пероксидные соединения хрома неустойчивы и быстро разлагаются с выделением кислорода и образованием ионов СгО2– (в щелочной среде) или Сr3+ (в кислой). Несколько более устойчив пероксид хрома в эфирном растворе. Реакцией его образования пользуются для открытия хрома. Обработкой аммиачного раствора хромата пероксидом водорода при 0 °С может быть получено коричневое пероксидное производное состава СrO4·3NН3, в котором хром, по-видимому, четырёхвалентен.
Пероксидные производные молибдена и вольфрама отвечают преимущественно типу M2ЭОn, где n изменяется от 5 до 8. Все они производятся от шестивалентных элементов и содержат в кислотном радикале от 1 до 4 пероксидных групп -О-О-, замещающих отдельные атомы кислорода. В частности, красный Nа2МоО8 может быть получен действием 30 %-ной Н2О2 на насыщенный раствор Nа2MoО4, при 0 °С. Соединение это при нагревании взрывается, а при комнатной температуре медленно отщепляет кислород, переходя в желтый Nа2МоО6, который разлагается со взрывом лишь при нагревании до 200 °С. В водном растворе пероксидные производные молибдена и вольфрама более или менее быстро разлагаются с отщеплением кислорода. Надвольфраматы, в общем, несколько устойчивее надмолибдатов.
При взаимодействии СrO3 и газообразного хлористого водорода образуется хлористый хромил (СrО2С12), представляющий собой красно-бурую жидкость. Соединения типа ЭО2Сl2 (при обычных условиях твёрдые) известны также для Mo и W. С водой все они взаимодействуют по схеме:
ЭО2Сl2 + 2 Н2О Ы ЭО2(ОН)2 + 2 НСl
В случае хрома равновесие практически нацело смещено вправо, т. е. хлористый хромил (подобно SO2С12) является типичным хлорангидридом. Производные Mo и W гидролизованы значительно меньше, что указывает на наличие у молибденовой и вольфрамовой кислот заметно выраженной амфотерности. В противоположность своим аналогам хлористый хромил является сильным окислителем.
Продукты полного замещения кислорода триоксидов ЭО3 на галогенид характерны только для Mo и W. Очень малые количества лимонно-желтого СrF6 образуются в качестве побочного продукта при нагревании хрома до 400 °С под высоким давлением фтора (350 атм). Из-за крайней неустойчивости этого вещества свойства его пока не изучены.
Фториды МоF6 и WF6 легко образуются путем непосредственного взаимодействия металлов с фтором. Для их теплот образования (в жидком состоянии) из элементов даются значения соответственно 1626 и 1722 кДж/моль. Оба они — МоF6 (т. пл. 18, т. кип. 35 °С) и WF6 (т. пл. 0, т. кип. 17 °С) — бесцветны, легкоплавки и очень летучи. В частности, несмотря на громадный молекулярный вес WF6 (почти 300), он при обычных условиях газообразен. Молекула WF6 представляет собой правильный октаэдр с атомом вольфрама в центре [d(WF) = 183 пм]. Интересно, что его растворы во многих органических жидкостях интенсивно окрашены. В противоположность SF6 рассматриваемые фториды весьма реакционноспособны. Водой они разлагаются с образованием бесцветных оксофторидов, из которых в свободном состоянии известны МоОF4 (т. пл. 97, т. кип. 196 °C), WOF4 (т. пл. 110, т. кип. 186 °С) и МоО2F2 (около 270 °С возгоняется). И для MoF6, и для WF6, были получены двойные соединения с фторидами щелочных металлов (К, Rb, Сs) типа, главным образом, ЭГ6·2МF. В безводной НF оба гексафторида растворяются, по-видимому, без химического взаимодействия.
Соединения типа ЭГ6 с другими галогенами известны для вольфрама. Темно-фиолетовый WСl6 (т. пл. 281, т. кип. 348 °С) образуется из элементов при нагревании (теплота образования 681 кДж/моль) и имеет структуру правильного октаэдра с вольфрамом в центре [d(WСl) = 224 пм]. Он хорошо растворим в спирте и эфире, практически не растворяется в воде на холоду, но легко разлагается ею при нагревании с образованием WOСl4 и WO2Сl2. Аналогичными свойствами обладает сине-чёрный WBr6, (теплота образования из элементов 385 кДж/моль). Имеется также сообщение о получении черного МоСl6, изоморфного WСl6 и неустойчивого в присутствии даже следов влаги.
Свободные кислоты типа НСrО3Г неизвестны, но их калийные соли были описаны для всех галогенов. Образуются они по общей схеме КГ+ СгО3 = КСrО3Г (в присутствии избытка соответствующей кислоты НГ). Водой эти галохроматы гидролизуются, но из достаточно кислых сред могут быть перекристаллизованы. Лучше других изучены красный фторохромат и оранжевый хлорохромат. Для молибдена и вольфрама наиболее характерны соли фторокислот, главным образом типов М2ЭО2F4, М2О3F2 и М3ЭО3F3, где М — одновалентный металл.
Отвечающие окcидам ЭО3сульфиды известны только для молибдена и вольфрама. При пропускании сероводорода в растворы молибдатов и вольфраматов происходит постепенное замещение кислорода серой с образованием соединений по ряду, например:
К2WO4 К2WO3S K2WO2S2 К2WOS3 K2WS4
бесцветный желтоватый желтый желтый красный
Для тетраэдрического иона WS42- имеем d(WS) = 217 пм.
Отвечающие всем перечисленным типам тиосоли легкорастворимы в воде. Поэтому в процессе пропускания Н2S осадка не образуется. Однако при сильном подкислении растворов рассматриваемые соединения разрушаются, например, по схеме:
К2ЭS4 + 2 HCl = Н2S + ЭS3 + 2 КСl
Практически нерастворимые в воде МоS3 и WS3 выделяются при этом в виде темно-коричневых осадков. Оба сульфида при нагревании на воздухе легко окисляются, а при прокаливании в отсутствие кислорода отщепляют серу и переходят в сульфиды ЭS2. Для молибдена были получены оба промежуточных оксосульфида — МоО2S и МоОS2. Сообщалось и о получении сульфохлорида вольфрама — WSСl4, (т. пл. 142 °С).
В качестве продуктов частичного восстановления изополисолей вольфрама (например, путем нагревания их в токе водорода) можно рассматривать т. н. вольфрамовые бронзы. Простейшей схемой образования этих веществ является взаимодействие вольфрамового ангидрида с металлическим натрием: хNа + WO3 = NаxWO3 (где 0
В кубических кристаллах вольфрамовых бронз, образованных радикалами WO3, имеются внутренние пустоты, меньшая или большая часть которых (в зависимости от величины х) заполняется ионами Nа+. Отщепляющиеся при переходах Nа ® Nа+ + е- валентные электроны натрия не присоединяются к каким-либо определенным атомам W+6 (переводя их в состояние W+5), а принадлежат решётке в целом, что характерно для металлов. Этим и обусловлено известное сходство вольфрамовых бронз с металлами.
В зависимости от состава окраска вольфрамовых бронз может быть различной, например, сине-фиолетовой (при х = 0,35), красной (при х = 0,62) или жёлтой (при х = 0,93), причём обычно она бывает очень красива. Это обстоятельство в сочетании с высокой устойчивостью по отношению к внешним воздействиям позволяет использовать вольфрамовые бронзы для изготовления высококачественных типографских красок. Помимо натрия, в их состав могут входить и другие металлы (Li, К, Rb, Cs, Тl, Са, Ва, Рb).
Изоструктурные вольфрамовым, тоже ярко окрашенные молибденовые бронзы МхМоО3 (где М — Nа, К, Rb) были синтезированы длительным взаимодействием М2МоО4, МоО3 и Mo при 400-1000 °С под давлением 60 тыс. атм. Частично они получены и методом электролиза. Примерами могут служить красная К0,26МоО3 и голубая К0,28МоО3. Отмечалось также получение Li-Mo бронз со степенью окисления молибдена 4,3ё5,8.
Изученные соединения пятивалентных элементов рассматриваемой группы сравнительно немногочисленны. Вольфрампентахлорид может быть получен повторной перегонкой WСl6 в токе водорода, молибденпентахлорид — нагреванием порошка Mo в токе хлора. И WСl5 (т. пл. 253, т. кип. 288 °С), и MоСl5 (т. пл. 194, т. кип. 268 °С) представляют собой зеленовато-чёрные кристаллические вещества. В твердом состоянии для них установлена димерная структура, но в парах они имеют строение тригональной бипирамиды [d(МоСl) = 227, d(WСl) = 226 пм]. Для обоих пентахлоридов получены зелёные двойные соединения типа МЭСl6 (где М — Cs др.), а для вольфрама и типа М2WСl7. Известны также желтый МоF5 (т. пл. 67, т. кип. 214 °С) и коричнево-фиолетовый WВr5 (т. пл. 276, т. кип. 333 °С). Тетрамерный в кристаллах WF5 неустойчив на воздухе, а около 60 °С дисмутирует по схеме: 2 WF5 = WF6 + WF4. Для него, как и для фторида молибдена, известны малоустойчивые двойные соединения с фторидами щелочных металлов (кроме лития) состава MЭF6 . Примером может служить NаМоF6 [d(МоF) = 174 пм].
Водой WСl5 и МоСl5 разлагаются по схеме:
ЭСl5 + Н2О = ЭОСl3 + 2 HСl
с образованием коричневого МоОСl3 или зелёного WOСl3. При одновременном наличии в растворе хлористых солей некоторых одновалентных металлов из него могут быть выделены зелёные двойные соединения, например WОСl3·2КСl и МоОСl3·2КCl. В виде неустойчивых на воздухе зелёных кристаллов была выделена и свободная кислота МоОСl3·2НСl. Отвечающие тому же типу ЭОГ3· 2МГ производные известны также для хуже изученных МоОF3, МоОВr3 и WOВr3. Выше 200 °С черный оксохлорид молибдена испаряется с частичным разложением по схеме:
3 МоОСl3 Ы МоОСl4 + МоО2Сl2 + МоСl3.
Восстановлением оксохлоридов ЭО2Сl2 хлористым оловом были получены МоО2Сl и WО2Сl.
При действии аммиака на растворы производных пятивалентного молибдена выпадает коричнево-бурый осадок MoО(ОН)3. Нагреванием его в токе углекислого газа может быть получен фиолетово-черный Мо2О5. Известен также Mo2S5, тогда как наличие аналогичных соединений у вольфрама не установлено. Из отвечающих Мо2О5 солей высокотемпературным синтезом были получены производные типа MII(МоО3)2 (где М = Мg, Са, Sr, Ва).
Частичное восстановление высших оксидов Mo и W ведет к образованию продуктов промежуточного между ЭО3 и Э2О5 состава. Большей частью они соответствуют формуле ЭxО3n-x (где х = 1, реже 2 или 3) и имеют синюю или фиолетовую окраску. Наиболее известным из таких смешанных продуктов восстановления оксидов ЭО3 является “молибденовая синь” (которой иногда приписывается определенная формула — Мо6О17).
Из производных пятивалентного хрома лучше других охарактеризованы соли типа М3СrО8 и темно-красные соли типа М2СrОСl5 (где М — К, Rb, Сs, NН4). Лежащий в их основе красновато-черный СrОСl3 был получен взаимодействием СrО3 с SOСl2. Он легко возгоняется в вакууме, но устойчив только при низких температурах. Известны и имеющие светлую пурпурную окраску соли типа МСrОF4 (где М — K и Аg). В форме двойного соединения СrОF3·0,3СlF3 выделен и соответствующий им красный оксофторид. Водой эти вещества тотчас разлагаются на производные CrIII и CrVI. Сине-чёрные соли Ва и Sr типа Э3(СrO4)2, получены сплавлением соответствующих хроматов и гидроксидов при 800 °С в токе азота по реакции:
4 ЭСrO4 + 2 Э(ОН)2 = 2 Э3(CrO4)2 + O2 + 2 Н2О
Сухим путём был получен и ряд других солей Н3СrО4 (большей частью имеющих зелёный цвет). Для иона СrO43-даются значения d(СrО) =167 пм. Под действием воды или кислот происходит дисмутация этих гипохроматов с образованием производных трёх- и шестивалентного хрома.
По-видимому, пятивалентен хром и в оксиде Сr2О5. Огненно-красный СrF5 является основным продуктом взаимодействия элементов под высоким давлением. Водой он быстро разлагается, образуя желто-зеленый раствор.
Из производных четырёхвалентного хрома его черный диоксид (СrО2) может быть, по-видимому, получен не только термическим разложением СrО3, но и нагреванием Сr(ОН)3 на воздухе (не выше 400 °С). Для синтеза чистого СrО2 рекомендуется длительное нагревание СrО2Сl2, приблизительно до 400 °С под давлением кислорода в 15-20 атм. Высокотемпературным синтезом были получены отвечающие ей зелёные соли — Ва2СrО4, Ва3СrО5 и Sr2СrO4. Водой они разлагаются на производные СrIII и СrVI. Взаимодействием СrО3 с расплавленным металлическим натрием был получен Nа2СrО3. Зеленовато-чёрный СrF4 образуется при нагревании до 300-350 °С порошка хрома в медленном токе фтора. Он плавится около 200 °С и при этой температуре заметно летуч (с образованием синего пара), расплывается во влажном воздухе и разъедает стекло. Известны также отвечающие ему двойные соединения типов CrF4·2МF и СrF4·МF, где М — К, Rb, Сs. Взаимодействие СrСl3 с хлором при 700 °С ведет к образованию СгСl4, который устойчив только в газовой фазе. В аналогичных условиях способен, по-видимому, существовать и СrВr4.
При взаимодействии СrО3 с иодатами одновалентных металлов образуются соединения MСrIO6. Исходя из состава, их можно было бы считать производными ортоиодной кислоты и четырехвалентного хрома. Однако, изучение строения показало, что в действительности они являются солями типа М[О3СrOIO2], т. е. производными смешанной иодатохромовой кислоты с шестивалентным хромом.
Диоксиды аналогов хрома — коричневые MoО2 и WO2 — образуются в качестве промежуточных продуктов при взаимодействии соответствующих металлов с кислородом и могут быть получены также восстановлением их высших окислов газообразным аммиаком. Они нерастворимы в воде и при нагревании на воздухе легко переходят в триоксиды. Теплота образования WO2 из элементов равна 589 кДж/моль.
Труднорастворимый и в щелочах, и в кислотах МоО2 довольно хорошо проводит электрический ток. Некоторые соли отвечающих ему кислот были получены высокотемпературным синтезом. Состав их выражается формулами К2МоО3, МIIМоО3 (где M — Сa, Sr, Ва) и М2IIМоO4 (где М — Ва или Sr).
Оснувной функции диоксидов отвечают галогениды четырехвалентных молибдена и вольфрама. Образующийся в результате взаимодействия МоО2 с хлором при нагревании в присутствии угля коричневый МоСl4 легко возгоняется в виде жёлтых паров. Его молекула представляет собой правильный тетраэдр с атомом Mo в центре и d(MoCl) = 223 пм. Аналогичную структуру имеет в парах и МоВr4 [d(МоВr) =239 пм]. Напротив, серо-бурый WСl4 нелетуч. Кристаллы его могут быть получены сильным нагреванием паров WСl6 в токе водорода. Они весьма гигроскопичны и разлагаются водой. И для хлорида, и для бромида известны двойные соединения с соответствующими галогенидами Сs, Rb, К — красные WСl4·2МСl и зелёные WВr4·2МВr. Аналогично WCl4 и WВr4 ведут себя по отношению к воде темно-зелёный WВгСl3 и черный WI4, тогда как красновато-коричневый WF4 значительно устойчивее. Напротив, светло-зелёный МоF4 легко гидролизуется. Значительно устойчивее по отношению к воде его двойные соединения, например темно-коричневый МоF4·2NаF. Для МоСl2 известны аналогичные по составу темно-зелёные двойные соединения с хлоридами Сs, Rb, К, а также коричневый оксохлорид МоОСl2, устойчивый по отношению к воде и соляной кислоте. Взаимодействием WO2 с НF при 500 °С был получен серый, весьма химически инертный оксофторид WОF2. Сообщалось также о получении WОСl2, WOВr2 и WOI2.
Из аналогичных по составу диоксидам серых сернистых соединений MоS2 встречается в природе и является важнейшей молибденовой рудой. Вольфрамдисульфид (WS2) может быть получен взаимодействием элементов при нагревании. Оба сульфида в воде нерастворимы и по отношению к ней устойчивы. При прокаливании на воздухе они сгорают с образованием соответствующих триоксидов. Молибдендисульфид плавится лишь при очень высоких температурах (около 2100 °С) и применяется иногда в качестве смазки для трущихся машинных частей, работающих под большими нагрузками.
Как уже отмечалось, из производных Cr, Mo и W в низших валентностях практически важнее других соединения трёхвалентного хрома. Сесквиоксид хрома (Сr2О3) легко образуется при прокаливании порошка металлического хрома на воздухе:
4 Сr + 3 O2 = 2 Сr2О3 + 2257 кДж
Он представляет собой очень тугоплавкое тёмно-зелёное вещество, нерастворимое не только в воде, но и в кислотах. Благодаря своей интенсивной окраске и большой устойчивости к атмосферным влияниям гемиоксид хрома служит прекрасным материалом для изготовления масляных красок (“хромовая зелень”).
В лабораторных условиях оксид хрома (т. пл. 2265 °С под давл.) удобно получать разложением бихромата аммония. Реакция по уравнению:
(NН4)2Сr2О7 = Сr2О3 + N2 + 4 Н2О + 514 кДж
начинается при нагревании до 280 °С, но дальше протекает самопроизвольно.
Если тот или иной оксид металла нерастворим в кислотах, его обычно переводят в растворимые соединения путём сплавления с каким-нибудь подходящим для этой цели веществом. Одним из последних является пиросульфат калия, распадающийся при высоких температурах с выделением SO3, который и действует на оксид металла, образуя с ним соответствующую сернокислую соль, например, по реакции:
Сr2O3 + 3 К2S2О7 = Сr2(SO4)3 + 3 К2SO4
Оксид хрома может быть переведен в растворимое соединение также сплавлением с селитрой и содой по реакции:
Сr2О3 + 3 NаNО3 + 2 Nа2СО3 = 2 Nа2СrO4 +3 NаNО2 +2 СО2)
или нагреванием с раствором NаВrО3 по реакции:
5 Сr2О3 + 6 NаВrО3 + 2 Н2О = 3 Nа2Сr2О7 + 2 Н2Сr2О7 + 3 Вr2
Как Cr2О3, так и отвечающие ей соли обычно получают не из металла, а путем восстановления производных шестивалентного хрома, например, по реакции:
К2Сr2O7 + 3 SO2 + Н2SO4 = К2SO4 + Сr2(SO4)3 + Н2O
Из солей сесквиоксида хрома (с катионом Сг3+) наиболее интересны хромовые квасцы — тёмно-фиолетовое кристаллическое соединение состава К2SО4·Cr2(SО4)3·24Н2О.
Действием NН4OН на раствор Сr2(SO4)3 может быть получен серо-синий осадок малорастворимого в воде гидрата гемиоксида хрома [Сr(ОН)3]. Последний имеет ясно выраженный амфотерный характер. С кислотами он дает соли трёхвалентного хрома, а при действии сильных щелочей — соли хромистой кислоты [НСrО2, т. е. Сr(ОН)3 — Н2О] с анионом СгО2-, называемые хромитами. Например:
Сr(ОН)3 + 3 НСl = СrСl3 + 3 Н2О
Сr(ОН)3 + КОН = КСrО2 + 2 Н2О
Таким образом, для растворённой части гидроксида хрома одновременно имеют место следующие равновесия:
Сr3+ + 3 OH-Ы Cr(ОН)3є Н3СrО3Ы НСrО2 + Н2ОЫН+ + СrO2- + Н2O
При добавлении кислот (Н+) эти равновесия смещаются влево, при добавлении щелочей (ОН+) — вправо.
Сама по себе электролитическая диссоциация Сr(ОН)3 и по тому и по другому направлению невелика, так как и основные, и особенно кислотные свойства гидроксида хрома выражены довольно слабо. Поэтому соли трехвалентного хрома подвергаются в растворах значительному гидролизу, а растворимые хромиты при отсутствии достаточного избытка щелочи гидролизованы практически нацело. Примером нерастворимого в воде хромита может служить природный хромистый железняк Fe(CrО2)2.
Если в кислой среде производные шестивалентного хрома легко восстанавливаются до наиболее устойчивых при этих условиях солей Сr3+, то в щелочной среде производные трехвалентного хрома довольно легко окисляются до хроматов свободными галогенами, пероксидом водорода и т. д., например, по реакциям:
2 КСrО2 + 3 Вr2 + 8 КОН = 6 КВr + 2 К2СrО4 + 4 Н2О
Сr2(SO4)3 + 3 Н2О2 + 10 NаОН = 3 Nа2SO4 + 2 Nа2СrО4 + 8 Н2О
Однако под действием кислорода воздуха окисление не идет.
Соли сесквиоксида хрома применяются главным образом в качестве протрав при крашении тканей и для хромового дубления кож. Большинство их хорошо растворимо в воде. С химической стороны эти соли интересны тем, что цвет их растворов меняется в зависимости от условий (температуры раствора, его концентрации, кислотности и т. д.) от зелёного до фиолетового. В частности, на холоду обычно наблюдается сине-фиолетовая окраска, а при нагревании — зелёная. Такое изменение окраски связано с различной гидратацией иона Сr3+. В кристаллическом состоянии большинство солей Сr3+ имеет фиолетовый цвет, но некоторые известны в обеих формах. Осаждение Сr(ОН)3 из их растворов под действием щелочей начинается при рН » 5,3. Полная константа основной диссоциации гидроксида хрома (по схеме Сr(ОН)3Ы Cr3+ + 3 ОН-) оценивается в 7·10-31, а константа первой ступени кислотной диссоциации по схеме:
Сr(ОН)3 Ы Н+ + СrО2- + Н2О
составляет 9·10-17.
Образующиеся при растворении Сr(ОН)3 в сильных щелочах хромиты могут производиться не только от НСrО2, но и от более богатых водой форм гидроксида хрома. Некоторые из солей последнего типа, например, зелёный Na3СrО3·3Н2О, были выделены. В растворе они способны существовать только при наличии большого избытка щелочи (выше 10 н.), а чистой водой легко разлагаются с выделением осадка Сr(ОН)3. Хромиты, получаемые сухим путем (сплавлением Cr2O3 с оксидами или карбонатами металлов), с точки зрения химической классификации производятся от НСrО2. Известны главным образом соли двухвалентных металлов; в воде они нерастворимы. Свободная НСrО2 представляет собой синевато-серое кристаллическое вещество, отщепляющее воду лишь около 430 °С. Потеря последних частиц воды сопровождается сильным саморазогреванием массы вследствие большого выделения тепла при кристаллизации образующейся Сr2О3.
Безводный хлорид хрома (III) СrСl3 образуется в результате взаимодействия элементов при нагревании (теплота образования 510 кДж/моль). Он представляет собой красно-фиолетовые кристаллы, довольно легко возгоняющиеся в токе хлора. В воде СrСl3 (т. пл. 1150 °С) сам по себе практически нерастворим. Однако в присутствии следов СrСl2 или какого-либо другого сильного восстановителя растворение идет быстро и со значительным выделением тепла. Упариванием образующегося раствора из него может быть выделен легкорастворимый в воде темно-зелёный кристаллогидрат СrСl3·6Н2О. Известен и фиолетовый кристаллогидрат того же состава. Тёмно-зелёная форма гидролизована значительно сильнее фиолетовой. С хлоридами СsёLi хромтрихлорид способен образовывать кристаллические двойные соединения, примером которых может служить розово-красный СrСl3·3КСl (т. пл. 838 °С). Теплоты их образования из исходных молекул последовательно возрастают по ряду (кДж/моль): -2,9 (Nа), +46,0 (K), 68,9 (Rb), 76,9 (Сs). Весьма близко к СrСl3 стоят по свойствам зелёный СrF3 (т. возг. 1200 °С), чёрные СrВr3 и СrI3.
Азотнокислый хром [Сr(NО3)3] образуется при растворении Сr(ОН)3 в азотной кислоте. Его раствор имеет в отражённом свете сине-фиолетовую, а в проходящем — красную окраску. При нагревании он зеленеет, но при охлаждении первоначальный цвет довольно быстро восстанавливается. Кристаллизуется Сr(NО3)3 в зависимости от условий с различным числом молекул воды.
Безводный сульфат хрома Сr2(SO4)3, подобно хлориду, растворяется в воде только при одновременном наличии следов какого-либо сильного восстановителя. Из раствора он выделяется обычно в виде фиолетового кристаллогидрата Сr2(SO4)3·18Н2О. Известны также более бедные водой зелёные кристаллогидраты, например Сr2(SO4)3·6Н2О. При совместной кристаллизации сернокислого хрома с сульфатами некоторых одновалентных катионов (Nа+, К+, Rb+, Сs+, NН4+, Тl+) выделяются тёмно-фиолетовые кристаллы хромовых квасцов, отвечающие составу: М2SO4·Сr2(SO4)3 24Н2О. Из них хорошо растворимые в воде (около 1:4 по массе при обычных условиях) калийные квасцы применяются в кожевенной промышленности. Помимо квасцов известно зелёное кристаллическое соединение сульфата хрома с серной кислотой состава Сr2(SO4)3·Н2SO4·14Н2О. Получены были и некоторые аналогичные производные селеновой кислоты.
Сульфат хрома Сr2S3 в водных растворах не образуется, но может быть получен сухим путем, например пропусканием сероводорода над раскаленным хлоридом хрома. Сернистый хром представляет собой черное кристаллическое вещество, практически нерастворимое в воде и лишь медленно разлагаемое ею. Отвечающий ему тиохромит натрия (NаСrS2) может быть получен сплавлением хромита с содой и серой. Известны также тиохромиты ряда двухвалентных металлов.
Для хрома довольно характерно образование некоторых хромит-хроматных производных. Так, при взаимодействии растворимых соединений СrIII и СrVI в нейтральной среде выпадает коричневый осадок хромитхромата [Сr(ОН)2]НСrO4, при длительном промывании водой переходящий в [Сr(ОН)]СrO4. Коричневый Сr2(СrO4)3, (т. е. суммарно Сr5О12) образуется в водном слое при извлечении пероксида хрома эфиром. Нагреванием смесей бихромата с хромовым ангидридом могут быть получены нерастворимые в воде чёрные соли щелочных металлов суммарного состава МCr3О8, отвечающие структуре МСr(СrO4)2.
Весьма характерным для производных Сr3+ является образование двойных соединений с аммиаком, примером которых может служить фиолетовый СrCl3·6NН3. Соединения эти в твёрдом состоянии довольно устойчивы, а водой частично разлагаются с выделением осадка Сr(ОН)3 по уравнению, например:
СrCl3·6NH3 + 6 H2O Ы Сr(ОН)3 + 3 NН4OН + 3 NН4Сl
Напротив, при наличии в растворе избытка аммиака и хлористого аммония равновесие смещается влево, т. е. в сторону растворения Сr(ОН)3 с образованием СrСl3·6NН3.
Рис. 5. Схема строения иона [W2Cl9]3–.
В противоположность хрому, для Mo и W трехвалентное состояние не характерно, и из относящихся сюда соединений известны лишь немногие. При нагревании МoCl5 до 250 °С в токе водорода образуется МоСl3, представляющий собой тёмно-красное кристаллическое вещество, нерастворимое не только в воде, но и в соляной кислоте. Аналогичные по составу чёрные бромид и иодид могут быть получены прямым синтезом из элементов, а желтоватый фторид — взаимодействием МоF5 с молибденом при нагревании. Его сплавлением с КF (в запаянной трубке при 800 °С) был получен бледно-жёлтый К3МоF6 (т. пл. 734 °С), анион которого представляет собой октаэдр с d(MoF) = 200 пм. Известны также оксогалогениды молибдена общей формулы МоОГ·4Н2О, где Г — F, Сl, Вr.
Хлорид трёхвалентного вольфрама известен в виде гексамера (WСl3)6 [с вероятной стпуктуро W6Сl12)Cl6] и в форме жёлто-зелёных двойных солей типа 2WСl3·3МСl, например 2WСl3·3КСl. Кристаллы этого соединения образованы катионами К и сложными анионами W2Cl93-, имеющими схематически показанную на рис. 5 пространственную структуру [d(WW) = 241, d(\WСl ) = 240, d(WСl ) = 218 пм]. Аналогичные по составу двойные соли были получены для МоСl3, МоВr3, МоF3 и CrCl3. Известна также более сложная по составу тёмно-зелёная двойная соль 3WСl3·5КСl, образующая при взаимодействии с водой тёмно-красный раствор. Сплавлением оксидов ЭО3 с СаСО3 при 1000 °С были получены Сa3МоО3 и Ca3WO3.
Отвечающий двухвалентному хрому хлористый хром (СrСl2) образуется при взаимодействии металла с соляной кислотой в атмосфере водорода. Он может быть также получен прокаливанием металического хрома в струе газообразного НСl или восстановлением СrСl3 водородом при температурах около 600 °С. Безводный CrCl2 представляет собой бесцветное кристаллическое вещество (т. пл. 824 °С), очень гигроскопичное и растворяющееся в воде с голубым окрашиванием раствора. Из последнего хлористый хром может быть выделен в виде синего кристаллогидрата CrСl2·4H2О, который выше 38 °С постепенно превращается в изомерную зелёную форму, а при 51 °С переходит в голубой тригидрат. Водный раствор зелёной формы тетрагидрата некоторое время сохраняет этот цвет и характеризуется значительно меньшей электропроводностью, чем равный по концентрации раствор синей формы тетрагидрата. От CrСl2 производятся некоторые двойные соли, примерами которых могут служить СrСl2·CsCl (т. пл. 709 °С) и СrСl2·2СsСl (т. пл. 563 °С с разл.).
Рис. 6. Схема строения Рис. 7. Схема строения иона Cr2(CH3COO)4·2H2O. [Mo6Cl8]4+.
Ион Сr2+ является настолько сильным восстановителем, что способен вытеснять водород из воды по схеме:
2Сr2+ +2 Н+ = 2 Сr3+ + Н2
(скорость этой реакции сильно зависит от каталитического влияния примесей). Кислородом воздуха он легко окисляется. Ввиду этого солянокислый раствор СrСl2 иногда применяют для поглощения кислорода; реакция идет по уравнению
4 СrСl2 + O2 + 4 НСl = 4 СrСl3 + 2 H2O.
Чёрный монооксид хрома (СrО) получить в индивидуальном состоянии очень трудно. Он весьма инертен химически, а при нагревании в вакууме выше 700 °С подвергается дисмутации на Сr2О3 и хром. Отвечающий ему гидрат Сr(ОН)2 выделяется в виде очень легко окисляющегося желтого осадка при действии щелочей на раствор СrСl2. Гидрат монооксида хрома имеет основной характер (К1·К2 = 1·10-17) и с кислотами образует соответствующие соли Сr2+. Из них наиболее устойчив малорастворимый ацетат хрома (II) Сr(СН3СОО)2·Н2О, выделяющийся в виде красного осадка при действии СН3СООNа на концентрированный раствор СrСl2. Он имеет димерную структуру, схематически показанную на рис. 6 [d(СrО) = 197, d(СrОН2) = 220, d(СrСr) = 246 пм]. С сернистым аммонием крепкий раствор CrСl2 дает черный осадок моносульфида хрома — СrS. От синего СrSО4·7Н2О производится также синяя двойная соль К2SO4·СrSO4·6Н2О. Сухим путем (прокаливанием металла в атмосфере галогеноводорода) были получены зеленый СrF2 (т. пл. 1100 °С), бесцветный CrВr2 (т. пл. 842 °С) и буро-красный СrI2 (т. пл. 795 °С). В противоположность труднорастворимому фториду, обе последние соли легкорастворимы в воде. Интересен смешанный фторид Сr2F5, содержащий одновременно атомы двух- и трёхвалентного хрома.
Из простых по составу производных двухвалентных Mo и W известны главным образом галогенидные соединения. Желтый МоСl2 может быть получен нагреванием молибдена в парах фосгена. Он практически нерастворим в воде, но растворяется в спирте и эфире. По данным рентгеноструктурного анализа, хлориду двухвалентного молибдена отвечает формула Мо6Сl12 со структурой [Мо6Сl8]Сl4. В катионе [Мо8Сl8] атомы хлора расположены по вершинам куба, а атомы молибдена — около середин его граней (рис. 7). Для ядерных расстояний даются значения: d(МоМо) = 260, d(СlСl) = 300 d(MoCl) = 250ё260 пм. При действии на Мо6Сl12 щелочей образуется основание [Мо6Cl8](OH)4, для которого получены соли некоторых кислот. С другой стороны, для Мо6Сl12, известны продукты присоединения типа Мо6Сl12·4КСl. Оранжевый бромид и черный иодид двухвалентного молибдена по свойствам аналогичны хлориду. Спеканием порошка молибдена с серой был синтезирован серый Мо2S3.
Серый, неустойчивый на воздухе WСl2 может быть получен нагреванием WСl4 в токе сухого диоксида углерода. Он является сильным восстановителем и при взаимодействии с водой энергично выделяет из нее газообразный водород. Те же свойства характерны и для зеленовато-жёлтого WВr2. Бурый WI2 в холодной воде практически нерастворим, а в горячей разлагается. Галогениды WГ2 могут быть получены также термическим разложением при 500 °С по схеме:
3 WГ4 = WГ2 + 2 WГ5
По-видимому, они подобны аналогичным соединениям молибдена (рис. 7), но производные ионов [W6Г8]4+ значительно менее устойчивы.
Особый интерес представляет ацетат двухвалентного молибдена — Mo(СH3СОО)2. Это желтое кристаллическое вещество настолько устойчиво, что около 300 °С возгоняется без разложения. Строение его подобно показанному на рис. 7, но без молекул воды [d(МоО) = 207ё212, d(МоМо) = 211 пм]. Известны аналогичные производные и некоторых других органических кислот.
При сопоставлении свойств серы и элементов обеих следующих за ней подгрупп наблюдается соответствие основных данных опыта учению об электронных аналогах: в производных высшей валентности аналогия серы с элементами подгруппы хрома выражена сильнее, чем с селеном и теллуром; напротив, в соединениях низших валентностей имеет место аналогия по ряду S-Sе-Те, тогда как члены подгруппы хрома теряют сходство с серой.
Действительно, кислоты Н2ЭО4 характерны для всех рассматриваемых элементов, но закономерное уменьшение их силы наблюдается лишь по ряду S-Сr-Mo-W (тогда как селеновая кислота по силе почти равна серной). Характерные для серы и членов подгруппы хрома соединения типа ЭО2Сl2 для селена и теллура не существуют.
С другой стороны, характерные для S, Sе и Те водородные соединения ЭН2 не имеют себе подобных в подгруппе хрома. Точно так же сходны у S, Sе и Те оксиды ЭО2 и производные от них кислоты Н2ЭО3, тогда как для элементов подгруппы хрома соответствующие оксиды малохарактерны (и имеют скорее основные свойства).
Первая группа периодической системы
Структура внешних электронных слоев в атомах элементов I группы позволяет прежде всего предполагать отсутствие у них тенденции к присоединению электронов. С другой стороны, отдача единственного внешнего электрона, казалось бы, должна происходить весьма легко и вести к образованию устойчивых одновалентных катионов рассматриваемых элементов.
Как показывает опыт, предположения эти в полной мере оправдываются только применительно к элементам левого столбца (Li, Na, K и аналогам). Для меди и ее аналогов они верны лишь наполовину: в смысле отсутствия у них тенденции к присоединению электронов. Вместе с тем их наиболее удалённый от ядра 18-электронный слой оказывается еще не вполне закреплённым и при известных условиях способен к частичной потере электронов. Последнее обусловливает возможность существования наряду с одновалентными Cu, Ag и Au также и соединений рассматриваемых элементов, отвечающих их более высокой валентности.
Подобное расхождение выведенных из атомных моделей предположений и результатов опыта показывает, что рассмотрение свойств элементов на основе только электронных структур атомов и без учёта остальных особенностей не всегда достаточно для химической характеристики этих элементов даже в самых грубых чертах.
Щелочные металлы
Применяемое к элементам ряда Li-Cs название щелочные металлы связано с тем, что их гидроксиды являются сильными щелочами. Натрий и калий относятся к наиболее распространенным элементам, составляя соответственно 2,0 и 1,1% от общего числа атомов земной коры. Содержание в ней лития (0,02%), рубидия (0,004%) и цезия (9·10-5) уже значительно меньше, а франция - ничтожно мало.
Хотя некоторые соединения натрия и калия (поваренная соль, сода, поташ) были известны еще в глубокой древности, различие между обоими элементами впервые установлено лишь в начале ХVIII века. Элементарные Na и K выделены только в 1807 г. Литий открыт в 1817 г., цезий и рубидий — соответственно в 1860 и 1861 г. Элемент № 87 — франций — был открыт в 1939 г., а название своё получил в 1946 г.
Природные натрий и цезий являются “чистыми” элементами (23Na и133Сs), литий слагается из изотопов 6Li (7,4%) и 7Li (92,6%), калий — 39K (93,22%) 40K (0,01%) и 41K (6,77%), рубидий — 85Rb (72,2%) и 87Rb (27,8%). Из изотопов франция основное значение имеет встречающийся в природе 223Fr (средняя продолжительность жизни атома 32 мин).
И ядро атома цезия, и его валентный электрон обладают магнитным моментом. Эти моменты могут быть взаимно ориентированы параллельно или антипараллельно. Разность энергий обоих состояний соответствует излучению со строго определенной частотой колебаний n = 9 192 631 770 с-1 (т. е. длиной волны l » 3,26 см). На этой основе были сконструированы цезиевые “атомные часы”, точность хода которых еще выше, чем у “молекулярных”.
В природе встречаются только соединения щелочных металлов. Натрий и калий являются постоянными составными частями многих силикатов. Из отдельных минералов натрия важнейший — поваренная соль (NaCl) входит в состав морской воды и на отдельных участках земной поверхности образует под слоем наносных пород громадные залежи каменной соли (Соликамск, Артемьевск, Илецк и т. д.). Верхние слои подобных залежей иногда содержат скопления солей калия в виде пластов сильвинита (mKCl·nNaCl), карналита (KCl·MgCl2·6H2O) и др., служащие основным источником получения соединений этого элемента. Имеющих промышленное значение природных скоплений калийных солей известно лишь немного. Важнейшее из них является Соликамское месторождение.
Природные залежи легкорастворимых солей на территории России образовались в результате постепенного усыхания внутренних морей, покрывавших в минувшие эпохи большую часть Европы и Западной Сибири. По запасам калия Соликамское месторождение превосходит все месторождения других стран мира, вместе взятые. Среднее содержание калия (по общепринятому расчёту на К2О) в сильвинитных пластах составляет около 15 %, в карналитовых — около 12 %. В составляющих продолжение Соликамских мощных соляных залежей Березниковского района попадаются пласты сильвинита с содержанием до 35 % К2О, а калийные месторождения южного Урала характеризуются значительным содержанием сернокислых солей (минералы каинит — KCl·MgSO4·3H2O, полигалит — K2SO4·MgSO4·2CaSO4·2H2O и др).
В противоположность калийным залежам природные источники получения солей натрия (моря, солёные озёра, каменная соль) имеются во многих странах. Мировой океан содержит 4·1015 т NaCl (а из каждой тысячи тонн морской воды практически получается около 1,3 т соли). Интересна громадная мощность некоторых месторождений каменной соли. Так, в Илецке ее непрерывный пласт разведан на глубину в полтора километра, причем никаких признаков приближения его нижней границы обнаружено не было.
Каменная соль непроницаема для не растворяющих ее жидкостей и сжатых газов. Пробурив отверстие в толщу ее месторождения и размыв (путем нагнетания воды и откачки раствора) достаточно большой свободный объём, можно затем использовать его в качестве подземного хранилища газа, нефти и т. д.
Следы NaCl (от 10-8 до 10-5 г/л) постоянно содержатся в атмосфере. Они попадают туда при испарении брызг морской воды. Было вычислено, что только с поверхности моря в атмосферу поступает несколько тысяч тонн соли за сутки. У 30 % взятых на высоте 1500 м облачных капель с радиусом > 5 мк ядра (с массой 10-12-10-13 г) оказались состоящими в основном из NaCl. Частицы соли были обнаружены также в кристаллах снега.
Для лития известен ряд минералов, но скопления их редки. Рубидий и цезий встречается почти исключительно в виде примесей к калию. Следы франция всегда содержатся в урановых рудах.
Минералами лития являются сподумен LiAl(SiO3)2 и лепидолит Li2KAl[Si4O10(F,OH)2]. Часть калия в последнем из них иногда бывает замещена на рубидий. То же относится к карналиту, который может служить хорошим источником получения рубидия. Для технологии цезия наиболее важен сравнительно редкий минерал поллуцит — CsAl(SiO3)2.
Соединения натрия имеют огромное значение для жизни. Достаточно напомнить хотя бы то, что человек потребляет ее ежегодно в среднем 5 кг NaCl. Подобным же образом для растений необходимы соли калия. В связи с этим около 90 % всех добываемых калийных соединений употребляется для удобрения почв. Остальные 10 %, равно как и громадные количества различных соединений натрия, используются в промышленности. Лишь сравнительно небольшое по объему применение находит пока производные лития и весьма ограниченное — соединение Rb и Cs.
Натрий у животных сосредоточен преимущественно в тканевых соках (лимфе крови), а калий — в самих тканях. Особенно богаты калием некоторые внутренние органы — печень, селезёнка и др. В целом взрослые животные организмы содержат обычно несколько больше калия, чем натрия (по массе). Напротив, в зародышах животных натрия гораздо больше, чем калия, причем соотношение между обоими элементами приближается к характерному для морской воды. Это можно рассматривать как непосредственное доказательство происхождения наземных животных из морских организмов.
Рубидий накапливается у растений главным образом в листьях, а у животных — в мышцах, несущих большую нагрузку (сердечной, грудной у птиц). Соединения лития и цезия токсичны. Вместе с тем наметилась возможность эффективного использования соединений лития при лечении некоторых психических заболеваний.
В организме взрослого человека имеется приблизительно 5 л крови, которая содержит около 0,6 % NaCl. Ежедневное выделение хлористого натрия с мочой составляет обычно около 15 г. Он выделяется из организма также с потом (который содержит около 0,5 % NaCl). При усиленном потоотделении (в горячих цехах и т. д.) для утоления жажды рекомендуется газированная воды, содержащая 0,5 % NaCl. Напротив, при гипертонии (повышенном кровяном давлении) рекомендуется ограничивать потребление поваренной соли.
Опытами на собаках было установлено, что при перегреве организма естественное равновесие между Na• и K•нарушается и возникает “калиевый голод”. Это указывает на целесообразность подсаливая потребляемой при усиленном потоотделении воды не просто поваренной солью, а смесью NaCl и KСl (вероятно, в соотношении около 10:1).
Насколько велико потребление калия растениями показывают следующие данные (в кг К на тонну):
| Озимая рожь | Яровая пшеница | Картофель | Сахарная свекла | ||||
| зерно | солома | зерно | солома | клубни | ботва | корни | ботва |
| 5,0 | 8,3 | 5,0 | 6,2 | 5,0 | 7,0 | 2,1 | 4,1 |
В результате снятия урожая по всему миру из почвы извлекается ежегодно более 25 млн. т калия и отдельные участки ее поверхности могут довольно быстро начать испытывать калийный голод. Обогащение почвы калием сопровождается в подобных случаях резким повышением урожайности. Особенно большое значение имеют калийные удобрения для таких важных культур, как картофель и сахарная свекла. Ежегодное мировое их производство (в пересчете на K2O) составляет около 20 млн. т. Содержание калия в обычном животном удобрении — навозе — составляет около 6 кг/т. С мочой человека ежедневно выделяется 2 г калия.
В противоположность легко вымываемым из почвы солям натрия соединения калия ею прочно удерживаются (за счёт сорбции глинами). Обстоятельство это имеет громадное значение для развития наземной растительности. С другой стороны, оно обусловливает относительную бедность природных вод солями калия. Например, воды нижнего течения Волги содержат калия в 25 раз меньше, чем натрия. В результате судьба обоих элементов на земной поверхности складывается прямо противоположно: тогда как соединения натрия концентрируются в морях, основным направлением геохимической истории калия является рассеивание его соединений в почве.
По естественному содержанию калия отдельные почвы различны: относительно много его в глинистых, мало в песчаных и торфянистых. Из природных форм нахождения этого элемента главной является ортоклаз, гораздо меньшее значение имеют мусковит, нефелин и др. Лишь около 1 % всего калия почва содержит в сорбированном состоянии, причем только десятая часть его водорастворима. Усвоение содержащегося в минералах калия растениями сильно затруднено (особенно в случае ортоклаза). Несколько облегчается оно тем, что главной составной частью корневых выделений является углекислый газ, благодаря чему непосредственно прилегающий к корням слой почвы имеет кислую реакцию (рН » 4). Всё же основное значение для питания растений (особенно — молодых) имеет не минеральный, а сорбированный калий.
Отдельные виды растений, помимо калия, избирательно извлекают и соединения других щелочных металлов. Так, некоторые солончаковые растения и морские водоросли содержат большое количество натрия. Литий накапливается в некоторых сортах табака, рубидий — в некоторых сортах свеклы. Рубидий всегда содержат также виноградные вина (в среднем: белые — 0,5 мг/л, красные — 1,1 мг/л).
В свободном состоянии щелочные металлы могут быть выделены электролизом их расплавленных хлористых солей. Основное практическое значение имеет натрий, ежегодная мировая выработка которого составляет более 500 тыс. т.
До введения в практику электролитического метода металлический натрий получали прокаливанием соды с углем по реакции:
Na2CO3 + 2 C + 1020 кДж = 3 CO + 2 Na
Ванна для получения металлического натрия электролизом хлорида натрия состоит из стального кожуха с шамотной футеровкой, графитовым анодом и кольцевым железным катодом, между которыми расположена сетчатая диафрагма. Электролитом служит обычно не чистый хлорид натрия (т. пл. 800 °С), а более легкоплавкая смесь из приблизительно 40% NaCl и 60% CaCl2, что дает возможность работать при температурах около 580 °С. Собирающийся в верхней части кольцевого катодного пространства и переходящий в сборник металлический натрий содержит небольшую (до 5%) примесь кальция, который затем почти полностью выделяется (растворимость Са в жидком натрии при температуре его плавления равна лишь 0,01%). По мере хода электролиза в ванну добавляют хлористый натрий. Расход электроэнергии составляет 15 кВт·ч на 1 кг натрия.
Выработка металлических калия и лития несравненно меньше, чем натрия. Литий получают электролизом расплава LiCl + KСl, а калий — действием паров натрия на расплав КСl, поступающий противотоком к ним в специальных дистиллированных колоннах (из верхней части которых выходят пары калия). Рубидий и цезий в больших масштабах почти не добываются. Для получения небольших количеств этих металлов удобно пользоваться нагреванием в вакууме их хлоридов с металлическим кальцием.
При отсутствии воздуха литий и его аналоги представляют собой серебристо-белые (за исключением желтоватого цезия) вещества с более или менее сильным металлическим блеском. Все щелочные металлы характеризуются небольшими плотностями, малой твёрдостью, низкими температурами плавления и кипения и хорошей электропроводностью. Их важнейшие константы приведены ниже.
| Li | Na | K | Rb | Cs | |
Плотность, г/см3 | 0,53 | 0,97 | 0,86 | 1,53 | 1,87 |
| Твёрдость (алмаз = 10) | 0,6 | 0,5 | 0,4 | 0,3 | 0,2 |
| Теплоёмкость (вода = 1) | 0,84 | 0,29 | 0,17 | 0,08 | 0,05 |
| Электропроводность (Нg = 1) | 11 | 21 | 14 | 8 | 5 |
Температура плавления, °С | 180 | 98 | 63 | 39 | 29 |
Температура кипения, °С | 1350 | 900 | 776 | 686 | 670 |
Франций в элементарном состоянии не получен. По химическим свойствам очень похож на рубидий и цезий.
Температуры плавления щелочных металлов с увеличением внешнего давления последовательно возрастают и приблизительно выравниваются. Так, при 30 тыс. атм у Li, Na, K и Rb они соответственно равны 234, 248, 251 и 267 °С.
В парах щелочные металлы главным образом одноатомны (содержание молекул Э2 составляет лишь несколько процентов). Обусловлено это малой устойчивостью двухатомных молекул, что видно из сопоставления их энергий диссоциации (при 25 °С):
Li2 | Na2 | K2 | Rb2 | Cs2 | |
| Ядерное расстояние, пм | 267 | 308 | 393 | 432 | 455 |
| Энергия диссоциации, кДж/моль | 107 | 72 | 49 | 45 | 43 |
Как и у галогенов, молекула Э2 тем устойчивее, чем меньше ядерное расстояние.
Если бы из молекул Э2 могла образоваться кристаллическая решётка, ее устойчивость обеспечивалась бы лишь слабыми межмолекулярными силами. Энергия атомизации такой решётки (по расчетам на моль) не очень отличалась бы от 1/2 энергии связи в самих молекулах. Сопоставление этих величин с действительными энергиями атомизации лития и его аналогов наглядно показывает, что металлическая структура для них гораздо (примерно в три раза) энергетически выгоднее молекулярной. Хотя d(ЭЭ) в металле существенно больше, чем в молекуле, и каждая отдельная связь Э-Э соответственно ослаблена, число связей атома с его ближайшими соседями в металле — 8 — гораздо больше, что и увеличивает общую энергию взаимодействия.
Пары щелочных металлов окрашены в характерные цвета: натрий — в пурпурно-красный, калий — в фиолетовый, рубидий — в оранжевый. Характерно окрашены также коллоидные растворы этих металлов (например, натрий в эфире имеет окраску от пурпурно-фиолетовой до синей, а калий — синевато-зелёную).
Внешне проявляющееся в виде окрашивания пламени испускание нагретыми атомами щелочных металлов световых лучей обусловлено перескоком электронов с наиболее высоких на более низкие энергетические уровни. Например, характерная жёлтая линия спектра натрия (слагающаяся из волн с длинами 589,0 и 589,6 нм) возникает при перескоке электрона с уровня 3р на уровень 3s. Очевидно, что для возможности такого перескока необходимо предварительное возбуждение атома, т. е. перевод одного или нескольких электронов на более высокий энергетический уровень. В рассматриваемом случае возбуждение достигается за счёт теплоты пламени (и требует затраты 201 кДж/моль), вообще же оно может последовать в результате сообщения атому энергии различных видов. Другие щелочные металлы вызывают появление следующих окрасок пламени: литий — карминово-красной (670,8 нм), калий — фиолетовой (404,4 нм), рубидий — синевато-красной (420,2 нм), цезий — синей (455,5 нм).
Благодаря малой плотности Li, Na и K всплывают на воде (Li — даже в керосине). Щелочные металлы легко режутся ножом, а твёрдость наиболее мягкого из них — цезия — не превышает твёрдость воска. Несветящееся пламя газовой горелки щелочные металлы и их летучие соединения окрашивают в характерные цвета, из которых наиболее интенсивен присущий натрию ярко-желтый.
Спектр люминесценции ночного неба показывает постоянное наличие в ней желтого излучения натрия. Высота места его возникновения оценивается в 200-300 км, т. е. атмосфера на этих высотах содержит атомы натрия ( конечно, в ничтожных количествах). Возникновение излучения описывается рядом элементарных процессов (звездочкой показано возбужденное состояние; М — любая третья частица — О2, О, N2 и др):
Na + O + M = NaO + M*, затем NaO + O = O2 + Na*
и, наконец, Na* = Na + hv.
В лучах Солнца атомы натрия испытывают прямое фотохимическое возбуждение и интенсивно светятся характерным для них желтым светом. Это было использовано для создания “искусственной кометы” путем испарения заключённого в ракете натрия (за счёт тепла сгорания термита) на высоте 150 тыс. км над Землёй. Образовавшее облако паров натрия за 4 мин достигло в поперечнике 600 км и было доступно непосредственному наблюдению. Его характеристики (интенсивность свечения, скорость рассеивания и др.) дали важные указания на некоторые особенности верхней атмосферы.
Так как у лития и его аналогов работа выхода электрона с поверхности металла невелика. такой выход может происходить под действием освещения их видимым светом (за счёт энергии поглощаемых фотонов). На этом явлении, которое носит название фотоэлектрического эффекта, основана работа фотоэлементов.
Относительная фотоэлектрическая чувствительность отдельных щелочных металлов к различным длинам волн видимого света различна. По степени восприятия различных цветов спектра наиболее приближается к человеческому глазу цезий.
Чувствительность человеческого глаза по отношению к свету различных цветов очень различна (максимальна она для l = 555 нм). Обстоятельство это весьма важно, так как более 85 % всех впечатлений человека имеет зрительное происхождение. Установлено, что глаз гораздо быстрее утомляется при восприятии красных и синих цветов, что под действием красного цвета внутриглазное давление повышается, а под действием зелёного снижается (по сравнению с обычным белым светом), что синий цвет действует успокаивающе на некоторых психологических больных (страдающих манией преследования). Оказалось, что при освещении электролампами из избирательно задерживающего жёлтые лучи стекла все предметы выглядят гораздо более чёткими. Было показано также, что производительность труда повышается до 25 %, если стены производственных помещений и различные находящиеся в них предметы имеют разную окраску. Все эти особенности цветного восприятия человека начинают серьезно учитываться при разработке проблем промышленной эстетики.
Область цветного восприятия животных может быть существенно иной, чем у человека. Например, пчела хорошо видит в ультрафиолетовом свете и различает другие цвета спектра, кроме красного (который кажется ей чёрным), а кошка вообще не различает цвета, и все они представляются ей лишь различными оттенками серого (как в обычном телевизоре). Сообщалось, что жёлтый цвет отпугивает акул.
Развитие растений стимулируется красным освещением и угнетается синим. Вместе с тем на культуре лука было сделано интересное наблюдение: оказалось, что красное освещение способствует повышению содержания углеводородов, а синее — белков.
По химическим свойствам щелочные металлы исключительно реакционноспособны (причем активность их по направлению от лития к цезию обычно возрастает). Во всех своих соединениях они одновалентны. Располагаясь в крайней левой части ряда напряжений, они энергично взаимодействуют с водой по схеме:
2 Э + 2 Н2О = 2 ЭОН + Н2.
При реакции с Li и Na выделение водорода не сопровождается его воспламенением, у К оно уже происходит, а у Rb и Cs взаимодействие протекает во взрывом.
В соприкосновении с воздухом свежие разрезы Na и K (в меньшей степени Li) тотчас покрываются рыхлой плёнкой продуктов окисления. Ввиду этого Na и K хранят обычно под керосином. Нагретые на воздухе Na и K легко загораются, а рубидий и цезий самовозгораются уже при обычной температуре.
При хранении металлического калия в соприкосновении с воздухом поверхность его постепенно покрывается более или менее толстым слоем пероксида (с промежуточной прослойкой из оксида). Пользование таким окислившимся калием (а также рубидием и цезием) иногда влечёт за собой сильные взрывы, обусловленные возникновением непосредственного контакта между пероксидом и металлом (например, при его разрезании). В случае натрия подобная опасность не грозит, так как при обычных условиях он окисляется только до оксида.
Хранить натрий и калий следует в плотно закрытых сосудах под слоем сухого и нейтрального керосина). Недопустим их контакт с кислотами, водой, хлорированными органическими соединениями (CСl4 и т. п.) и твердым диоксидом углерода. Нельзя накапливать мелкие обрезки калия, которые окисляются особенно легко (из-за своей относительно большой поверхности). Неиспользованные остатки калия и натрия при малых их количествах уничтожают взаимодействием с избытком спирта, при больших — сжиганием на углях костра. Загоревшиеся в помещении щелочные металлы лучше всего тушить, засыпая сухим порошком кальценированной соды.
С рядом металлов (Ag, Au, Cd, Zn, Pb и др.) натрий сплавляется, тогда как с другими (Al, Fe, Ni, Cr, Mn и др.)сплавов не образует. Все щелочные металлы растворимы в ртути (хуже других — литий), причем с повышением температуры растворимость увеличивается.
Натрием широко пользуются при синтезах органических соединений и отчасти для получения некоторых его производных. В ядерной технике он используется как теплоноситель. Создающий яркий жёлтый свет электрический разряд в парах натрия является наиболее экономичным (но неприятным по сообщаемым им окружающим предметам оттенкам) источником искусственного освещения с коэффициентом полезного действия тока до 70 %. Проведены успешные опыты по созданию натриевого электрокабеля (с полиэтиленовой обкладкой) для токов высокого напряжения. В виде амальгамы натрий часто применяется как энергичный восстановитель.
Литий имеет совершенно исключительное значение для термоядерной техники. В резиновой промышленности он используется при выработке искусственного каучука (как катализатор полимеризации), в металлургии — как ценная присадка к некоторым другим металлам и сплавам. Например, присадка лишь сотых долей процента лития сильно повышает твёрдость алюминия и его сплавов, а присадка 0,4 % лития к свинцу почти в три раза повышает его твёрдость, не ухудшая сопротивления на изгиб. Имеются указания на то, что подобная же присадка цезия сильно улучшает механические свойства магния и предохраняет его от коррозии, однако такое его использование вряд ли вероятно из-за дороговизны металла: на мировом рынке (1960 г.) и цезий и рубидий расценивались в 7,5 раза дороже серебра.
Цезий применяется главным образом для изготовления фотоэлементов, а рубидий и его соединения пока почти не используются. Между тем скоро они будут получаться в больших количествах как один из продуктов переработки соликамских карналитов (содержащих 0,003-0,012 вес. % RbCl и около 0,0002 вес. % CsCl). Поэтому важной становится проблема изыскания рациональных путей ассимиляции рубидия.
Пары калия находят интересное использование в установках для прямого преобразования тепловой энергии в электрическую — магнитогидродинамических генераторах. Принцип их работы основан на том. что в пропускаемом с большой скоростью сквозь интенсивное магнитное поле сильно нагретом потоке частично ионизированного газа (“плазме”) возникает электрический ток. Так как пары калия сравнительно легко ионизируются, введение его соединений (например, K2CO3) в продукты сгорания топлива позволяют существенно повысить электропроводность плазмы при относительно низких температурах (порядка 2500 °С). Первый МГД — генератор мощностью 25 тыс. кВт уже работает в Москве.
Другой интересный путь возможного использования относительно легкой ионизируемости атомов щелочных металлов связан с проблемой ионного двигателя. Если ионизацией паров (например в электрической дуге) создать плазму, затем электрическим полем разделить ионы Э+ и электроны, разогнать их при помощи ускорителей и вновь соединить у выхода из сопла ракеты, то вылетающий поток атомов создаёт реактивную тягу. Последняя очень мала, но может быть использована уже находящейся в космическом пространстве ракетой для постепенного набора скорости или изменения траектории полёта. Подсчёты показывают, что расходующий 500 г цезия в час ионный двигатель способен обеспечить космическому кораблю с массой в 1 тыс. т. ускорение порядка 1 м/с2 и конечную скорость до 150 км/с. Источником энергии при этом должна быть атомная электростанция.
Жидкий в обычных условиях сплав (приблизительно 30-80 ат. % К) находит использование при органических синтезах. В лабораторных условиях его обычно готовят путем сдавливания очищенных от оксидных плёнок кусочков калия и натрия в фарфоровой ступке под слоем керосина (операция довольно опасна, так как взаимодействие сопровождается вспышками). В технике этим сплавом (заключённым в систему труб) пользуются иногда для быстрого переноса тепла. Интересно, что образование его сопровождается некоторым сжатием системы, но одновременно с этим ее сжимаемость не уменьшается, а возрастает. Также интересно, что при длительном пропускании сквозь жидкий сплав постоянного тока у анода накапливается не какой-либо один из двух металлов, а тот, которого в сплаве меньше. Вместе с тем результаты структурного исследования этого сплава говорят за наличие тенденции к образованию пар из разных атомов.
При наличии следов влаги щелочные металлы воспламеняются в атмосфере хлора. Взаимодействие Cs, Rb и K с жидким бромом сопровождается сильным взрывом, тогда как Na и Li при обычных температурах реагируют только поверхностно. С иодом реакции протекают подобным же образом, но менее энергично. Во всех случаях взаимодействия с галогенами продуктом реакции является соответствующая соль (ЭГ).
Образование сульфида Э2S при растирании щелочного металла с порошком серы сопровождается взрывом. При нагревании в атмосфере водорода литий и его аналоги образуют гидриды ЭH, имеющие характер типичных солей, в которых отрицательным ионом является водород H-. С азотом и углеродом непосредственно соединяется только литий. Образование его нитрида Li3N медленно идет в атмосфере азота уже при обычных температурах. Напротив, карбид лития Li2C2 может быть получен из элементов лишь при нагревании.
В соответствии с ходом изменения ионизационных потенциалов щелочных металлов можно было бы ожидать, что в ряду напряжений левее всех будет располагаться Сs, правее — Li. Наблюдаемое на опыте высокое значение нормального потенциала лития обусловлено большой энергией гидратации его положительного иона. Действительно, за счёт гидротации иона Э+ (по схеме: Э+ + aq Ы Э·) имеющее место у электрода равновесие Э Ы Э+ + е должно смещаться вправо, и тем больше, чем энергичнее данный ион гидратируется. Этим же обусловлено и выравнивание нормальных потенциалов тяжёлых щелочных металлов.
Щелочные металлы растворимы в жидком аммиаке и некоторых органических аминах. Из раствора лития в жидком аммиаке был выделен нейтральный аммиакат Li(NH3)4, аналогичный подобным же соединениям щелочноземельных металлов, из раствора натрия в пиридине — тёмно-зелёный комплекс Na(C5H5N)2. Интересна растворимость калия (но не натрия) в тетрагидрофуране, диглиме и некоторых других эфирах — образующиеся разбавленные (лишь около 10-4 моль/л) голубые растворы в отсутствии воздуха устойчивые. Аналогичный голубой раствор калия может быть при 0 °С получен и в воде (освобожденной от растворенного воздуха), но он неустойчив. Подобные системы, как и в случае жидкого аммиака содержат сольватированные катионы и поляроны.
В водном растворе калия таким поляроном является гидратированный электрон — е’. Возникновение его по схеме К+ + aq Ы K•+ e’связано с тем, что сумма теплот гидротации К+ (339 кДж/моль) и е’ (159 кДж/моль) практически равна сумме теплот атомизации калия (88 кДж/моль) и ионизации его атома (420 кДж/моль). При отсутствии других возможностей (в частности окислителей) гидротированный электрон взаимодействует с водой по схеме е’+ Н2О Ы Н + ОН’(энергия активации 21 кДж/моль), причем К = [H][OH’]/[e’][H2O] = 7·10-7. Кислая Среда смещает равновесие вправо, щелочная — влево. Однако из-за реакции Н + Н = Н2 оно быстро нарушается.
Довольно значительные концентрации гидратированных электроном могут быть получены пропусканием атомарного водорода в сильно щелочную среду (рН > 12). Атом водорода диссоциирует при этом по схеме Н + aq Ы H• + e’, т. е. ведёт себя как слабая кислота (К = 2·10-10).
Как и всякий полярон е’ представляет собой образование, в котором е поляризационно связан с частицами Среды. Предполагается, что электрон находится в тетраэдрическом окружении четырех молекул воды. Заряд его располагается, по-видимому, в области с радиусом 140 пм. Существует также предположение, что сольватируются не единичные электроны, а их пары (с антипараллельными спинами), т. е. раствор содержит ионы типа е2”. Наличие подобных сольватированных аммиаком электронных пар весьма вероятно для самих диамагнитных раствором натрия в жидком NH3.
Гидратированный электрон является чрезвычайно сильным восстановителем. Он способен восстанавливать некоторые ионы (Pb••, Cd••, Ni••, Co••, Cr•••, Zn••), не реагирующие с атомарным водородом и особенно активен по отношению к частицам с непарными электронами (NO и др.). Его взаимодействие с катионами идет тем быстрее, чем выше их заряды и больше радиусы, а с анионами — чем их заряды ниже.
Взаимодействие растворенного в жидком аммиаке щелочного металла с монооксидом углерода сопровождается образованием белых (или имеющих бледные цветные оттенки) солеобразных продуктов состава ЭСО. Строение их отвечает формуле Э2+[OCєCO]2-. Те же продукты могут быть получены прямым взаимодействием щелочных металлов (кроме Li) c монооксидом углерода при температурах ниже 230 °С (тогда как при более высоких температурах образуются соли гексаоксибензола — М6С6О6). Таким образом, рассматриваемые соединения являются в действительности не карбонилами щелочных металлов, а производными оксиацетилена (или гексаоксибензола).
Вещества эти гидроскопичны и пирофорны. Нагревание их в вакууме сопровождается разложением по схеме:
2 Э2С2О2 = Э2СО3 + Э2О + 3 С.
С водой они взаимодействуют бурно (вплоть до взрыва). При обработке белого Na2C2O2 водяным паром он становится красным, затем фиолетово-чёрным, после чего за несколько дней превращается в вязкую красную жидкость. Подобная же обработка черного К2С2О2 ведёт к его покраснению, а затем пожелтению. Первоначально жёлтый раствор К2С2О2 в большом количестве воды быстро краснеет. При упаривании он вновь желтеет. Из него были выделены тёмно-жёлтые кристаллы кроконата калия — К2С5О5 (кроконовая кислота представляет собой пятичленный цикл из трех групп СО и двух СОН с двойной связью между ними.
Образование взрывчатого К2С2О2 может происходить также при прокаливании смеси поташа с углем. Поэтому для получения металлического калия такой метод непригоден.
При действии СО2 на осажденный в вакууме тонкий слой цезия образуется синее вещество состава Cs2CO2. Так как оно гидролизуется по схеме:
CsCO2 + H2O = CsOH + HCOOCs,
строение его должно отвечать формуле CsCOOCs (т. е. оно может рассматриваться как продукт замещения на цезий обоих атомов водорода муравьиной кислоты). Нагреванием под вакуумом сопровождается частичным отщеплением цезия с образованием его оксалата:
2 CsCOOCs = 2 Cs + Cs2C2O4.
Металлический цезий способен присоединять этилен с образованием твердого коричневого продукта С2Н4Cs2. Водой это соединение разлагается на С2Н6 и CsOH. В реакцию с бензолом цезий медленно вступает уже при обычных температурах, образуя черный осадок С6Н5Cs (который на воздухе самовоспламеняется). Рубидий реагирует подобным же образом, но лишь при 70 °С. Другие щелочные металлы с бензолом не взаимодействуют.
При сгорании щелочных металлов в избытке кислорода образуются соединения следующего состава и цвета:
Li2O Na2O2 KO2 RbO2 CsO2
белый белый жёлтый жёлтый жёлтый
Из всех этих веществ нормальным оксидом является только Li2O, а остальные представляют собой пероксидные соединения.
Практическое применение находит главным образом пероксид натрия (Na2O2). Технически ее получают окислением при 350 °С распыленного металлического натрия:
2 Na + O2 = Na2O2 + 510 кДж.
Образующийся продукт обычно представляет собой порошок или крупинки желтоватого цвета.
Взаимодействие Na2O2 c водой сопровождается гидролизом:
Na2O2 + 2 H2O Ы 2 NaOH + H2O2 + 142 кДж.
На выделении Н2О2 при этой реакции основано использование пероксида натрия для отбелки различных материалов. Взаимодействие Na2O2 c диоксидом углерода по схеме:
2 Na2O2 + 2 CO2 = 2 Na2CO3 + O2 + 464 кДж
служит основой применения пероксида натрия как источника кислорода в изолирующих противогазах и на подводных лодках. С легко окисляющимися веществами пероксид натрия реагирует настолько энергично, что взрыв может иногда последовать уже при простом соприкосновении.
Чистый Na2O2 бесцветен, но поступающий в продажу препарат обычно имеет желтую окраску (из-за примеси около 10 % NaO2).
Термическое разложение пероксида натрия происходит по схеме:
2 Na2O2 = 2 Na2O + O2
начинает становиться заметным уже с 400 °С, а давление кислорода в одну атмосферу достигается при 636 °С. Под его избыточным давлением Na2O2 плавится при 600 °С.
При взаимодействии Na2O2 с водой происходит сильное разогревание, обусловленное образованием гидрата Na2O2·8H2O. Известно также кристаллическое соединение состава Na2O2·2H2O2·4H2O, теряющее воду при хранении в эксикаторе над серной кислотой. Аналогичное соединение калия кристаллизуется без воды. Оба вещества могут быть получены путем обработки соответствующих гидроксидов крепким пероксидом водорода при 0 °С.
При осторожной обработке пероксида натрия охлажденным до 0 °С спиртом по реакции:
Na2O2 + C2H5OH = C2H5ONa + NaOOH
в виде белого порошка осаждается кислая соль пероксида водорода. Вещество это — гидропероксид натрия — отдаёт кислород еще легче, чем Na2O2, а с диоксидом углерода образует NaHCO4.
Чистый или содержащий различные добавки (например, хлорной извести с примесью солей никеля или меди) пероксид натрия носит техническое название “оксилит”. Смешанные препараты оксилита особенно удобны для получения кислорода, который выделяется ими под действием воды. Спрессованный в кубики оксилит может быть использован для получения равномерного тока кислорода в обычном аппарате для получения газов.
При сжигании лития в токе кислорода наряду с Li2O образуется также небольшие количества пероксида лития — Li2O2. В индивидуальном состоянии он может быть получен взаимодействием кипящего раствора LiOH (2 г/л) с 30 % раствором Н2О2. Образующийся осадок состава Li2O2·H2O2·3H2O промывают спиртом и затем выдерживают под вакуумом над фосфорным ангидридом (что ведёт к потере и Н2О и Н2О2). Термическое разложение пероксида лития по схеме:
2 Li2O2 = 2 Li2O + O2
наступает около 300 °С.
Нехарактерные для К, Rb и Cs пероксиды Э2О2 могут быть получены в виде белых (или желтоватых) осадков действием точно рассчитанного количества кислорода на растворы соответствующих металлов в жидком аммиаке. Избытком кислорода они легко переводятся в надпероксиды ЭО2 (причем промежуточно образуются смеси Э2О2 и ЭО2 в том числе состава Э2О3). По окислительным свойствам все пероксиды Э2О3 других щелочных металлов похожи на пероксид натрия.
Характерные для K, Rb и Cs надпероксиды ЭО2 могут быть получены сжиганием металлов на воздухе [их теплоты образования из элементов практически одинаковы: 284 (K, Rb) или 288 (Cs) кДж/моль]. Они представляют собой твёрдые жёлтые вещества, кристаллические решётки которых подобны решётки СаС2.
Термический распад надпероксидов по схеме:
ЭО2® Э2О3® Э2О
начинает становиться заметным около 400 °С (по другим данным, при атмосферном давлении КО2 устойчив до 530 °С). С водой они реагируют по схеме:
2 ЭО2 + 2 Н2О = 2 ЭОН + Н2О2 + О2
(в случае К2О тепловой эффект равен 54 кДж/моль), а со способными окисляться веществами реакции протекают настолько бурно, что могут сопровождаться взрывом.
Надпероксид калия (КО2) нередко вводится в состав оксилита. Его взаимодействие с углекислым газом идет в этом случае по суммарному уравнению:
Na2O2 + 2 KO2 + 2 CO2 = Na2CO3 + K2CO3 + 2 O2 + 420 кДж,
т. е. диоксид углерода заменяется равным объемом кислорода.
Нагреванием Na2O2 до 400 °С под давлением кислорода в 150 атм может быть получен надпероксид натрия NaO2 аналогичный соответствующим производным K, Rb и Cs, но менее устойчивый и характеризующийся решеткой типа пирита с d(OO) = 133 пм. Теплота его образования из элементов равна 259 кДж/моль. Это жёлтый гигроскопичный порошок, быстро разлагающийся во влажном воздухе. При 100 °С надпероксид натрия взаимодействует с монооксидом углерода по уравнению:
2 NaO2 + CO = Na2CO3 + O2.
Аналогично идет реакция с диоксидом углерода при обычной температуре, но ниже 10 °С образуется надкарбонат:
2 NaO2 + 2 CO2 = Na2C2O6 + O2 NaO2.
При -80 °С цвет NaO2 меняется на белый, что сопровождается изменением также магнитных свойств.
Взаимодействие O3 с суспензией Li2O2 во фреоне-12 при -65 °С было получено жёлтое твёрдое вещество с содержанием до 45 % LiO2. Этот надпероксид способен существовать лишь ниже -35 °С. По строению он подобен надпероксиду натрия.
Кроме щелочных металлов надпероксиды известны только для элементов подгруппы кальция. В индивидуальном состоянии они не выделены, но разложением при определенных условиях пероксидных производных типа ЭО2·2Н2О2 были получены смеси состава хЭ(ОН)2·уЭО2·zЭ(O2)2 со следующим максимальным содержанием надпероксидов (вес. %): 40 (Ca), 30 (Sr) и 11 (Ba). При хранении вне контакта с воздухом они устойчивы, и с водой бурно взаимодействуют, отщепляя надпероксидный кислород. Из производных комплексных катионов получен устойчивый до 100 °С жёлтый надпероксид тетраметиламмония — [N(CH3)4]O2 (т. пл. 97 °С).
Лежащий в основе надпероксидов радикал гидропероксид НО2 способен существовать лишь ничтожные доли секунды, после чего распадается по схеме:
2 НО2 = Н2О2 + О2.
Однако некоторые его характеристики известны: теплота образования из элементов составляет 21 кДж/моль. Энергия связи Н-О2 оценивается в 196,5 кДж/моль.
Сочетание двух таких радикалов могло бы дать надпероксид водорода — Н2О4. Существует предположение, что она частично образуется в результате взаимодействия атомарного водорода с твердым озоном при -196 °С по схеме:
2 Н + 2 О3 = 2 НО2 + О2 = Н2О4 + О2.
Кроме рассматривавшихся выше пероксидных производных для Na, K, Rb и Cs уже давно были известны озониды. Вещества эти образуются в виде оранжево-красной корки на поверхности омываемых током озона твердых гидроксидов. Используя их растворимость в жидком аммиаке (например, до 15 г/100 г NH3 для соли калия), удается выделить озониды ЭО3 в более или менее чистом состоянии.
Образование лучше всего изученного озонида калия протекает по суммарной схеме:
4 KOH + 4 O3 = 4 KO3 + O2 + 2 H2O
(причем вода связывается избыточным КОН). Энергия активации этой реакции составляет лишь 12,5 кДж, а теплота образования КО3 из элементов равна 259 кДж/моль.
2 KO3 + 2 KO2 + O2 + 192 кДж/моль
уже в обычных условиях (быстро и нацело реакция протекает при +60 °С). Водой он мгновенно разлагается по суммарной схеме:
4 KO3 + 2 H2O = 4 KOH + 5 O2
(по-видимому, с промежуточным образованием радикалов ОН). Озонид калия является типичной солью, образованной ионами K+ и O3- [с параметрами d(OO) = 134 пм и РOOO = 100°]. Аналогичные свойства имеют и другие рассматриваемые озониды, причем устойчивость их по ряду Na-K-Rb-Cs возрастает. Так, NaO3 быстро распадается уже при -10 °С, а CsO3— лишь при 100 °С. Последняя соль была синтезирована взаимодействием CsO2 с озонированным кислородом. Для всех озонидов характерно сильное светопоглощение в области 400-500 нм.
Для лития озонид известен лишь в форме красного аммиачного комплекса [Li(NH3)4]O3 разлагающегося при отщеплении NH3. Получен также красный —NH4O3, уже выше -126 °С разлагающийся по уравнению:
4 NH4O3 = 4 H2O + 2 NH4NO3 + O2.
Гораздо устойчивее (до 25 °С) красный озонид тетраметиламмония — [N(CH3)4]O3.
Нормальные оксиды щелочных металлов (за исключением Li2O) могут быть получены только косвенным путем. Они представляют собой твёрдые вещества следующих цветов:
Li2O Na2O K2O Rb2O Cs2O
белый белый белый жёлтый оранжевый.
Оксид лития гидратируется сравнительно медленно. Напротив, оксиды остальных щелочных металлов реагируют с водой весьма энергично. Взаимодействие протекает по схеме:
Э2O + H2O = 2 ЭOH
и сопровождается большим выделением тепла.
Гидроксиды ЭOH щелочных металлов представляют собой бесцветные, очень гигроскопичные вещества, разъедающие большинство соприкасающихся с ними материалов. Отсюда их иногда употребляемое в практике название — едкие щелочи. Все они сравнительно легкоплавки и летучи без разложения (кроме отщепляющей воду LiOH).
В воде гидроксиды щелочных металлов хорошо растворимы (хуже других — LiOH), причем почти нацело диссоциированы на ионы Э• и OH’. Так как эта диссоциация больше, чем у гидроксидов всех других металлов, едкие щелочи являются самыми сильными основаниями.
Гидроксид натрия (иначе: едкий натр, каустическая сода) потребляется многими отраслями промышленности. Ее ежегодная мировая выработка исчисляется миллионами тонн, причем бульшая часть добывается электролизом растворов NaCl. Реже пользуются обменным разложением соды с гашеной известью:
Na2CO3 + Ca(OH)2 = CaCO3Ї + 2 NaOH
Реакция эта использовалась еще в древнем Египте.
Из гидроксидов других щелочных металлов значительное практическое применение находит только КОН («едкое кали»). Вырабатывают его обычно электролизом растворов КCl.
Получаемый сжиганием металла Li2O содержит примесь Li2O2. Чистый оксид лития может быть получен термическим разложением Li2СО3 (при 700 °С в вакууме).
Применительно к оксиду натрия наилучшие результаты дает взаимодействие в вакууме NаN3 с NаNО3, протекающее по реакции:
5 NаN3 + NаNО3 = 8 N2 + 3 Nа2О.
Оксиды К, Rb и Сs рекомендуется получать путем окисления расплавленных металлов недостаточным количеством кислорода с последующей отгонкой избытка металла в вакууме. Теплоты образования оксидов Э2О из элементов равны (кДж/моль): 598 (Li), 414 (Nа), 359 (К), 330 (Rb), 318 (Сs). Производные Li-Rb кристаллизуются по типу СdI2 (рис. Х11-37), а Сs2О — по типу СaF2 (рис. Х111-69) с обратным расположением катионов и анионов [d(ОСs) = 286, d(СsСs) = 419 пм]. Оксид лития входит в состав специальных стекол (10-24 % Li2O, 2-13 % ВеО, 70-83 % В2О3), прозрачных для рентгеновских лучей. При нагревании белый К2О желтеет, бледно-желтый Rb2О краснеет, а оранжевый Сs2О становится почти черным.
Для точек плавления и кипения оксида лития даются значения 1570 и 2600 °С, однако гораздо раньше начинается его испарение, сопровождавшееся частичной диссоциацией на элементы (степень которой при 1000 °С оценивается в 10 %). Молекула Li2О, по-видимому, линейна с d(LiO) = 160 пм. По ряду Li-Сs летучесть оксидов возрастает, Так, при давлении 10-5 мм рт. ст. Li2O до 980 °С испаряется очень незначительно, испарение Nа2О становится заметным около 670, К2О — около 430, а Сs2О — около 350 °С. Приблизительно при тех же температурах начинается дисмутация оксидов Nа, К, Rb, Сs по схеме 2 Э2О = Э2О2 +2 Э. Расчетным путем было показано, что выше 1800 °С натрий с кислородом не взаимодействует.
Некоторые свойства гидроксидов щелочных металлов сопоставлены ниже:
| LiOH | NaOH | KOH | RbOH | CsOH | |
Теплота образования из Э2О кДж/моль | 46,4 | 75,7 | 102 | 105,8 | 104,9 |
Плотность, г/см3 | 1,4 | 2,1 | 2,1 | 3,2 | 3,7 |
Энергия кристаллической решетки, кДж/моль | 857 | 736 | 640 | 614 | 568 |
Т плавления, °С | 473 | 321 | 405 | 382 | 346 |
Растворимость, моль/л Н2О при 15 °С | 5,1 | 15,9 | 19,2 | 17,9 | 25,8 |
при 20 °С | 5,2 | 29,8 | 22,5 | 16,9 | 20,2 |
| Теплота растворения, кДж/моль | 21,3 | 43,0 | 55,2 | 61,9 | 71,1 |
Расплавленные гидроксиды щелочных металлов имеют в основном ионную структуру. Так как они сильно разъедают стеклянную, фарфоровую и (при доступе воздуха) платиновую посуду, для их плавления пользуются сосудами из серебра, никеля или железа. Содержащая 50 мол. % каждого компонента система NаОН + КОН плавится при 170 °С.
Взаимодействие расплавленного гидроксида натрия со способными окисляться металлами идет в основном по схеме (для двухвалентного М)
2 NаОН + М = MO + Nа2О + Н2
причем практически нерастворимый в расплавленных щелочах водород уходит из сферы реакции. По такому типу реагируют, например, Тl, Та, Сr, Мn. В качестве вторичных реакций иногда могут фигурировать различные другие процессы, например
Nа2О + MO = Nа2MО2 (у Ве)
или Nа2О + М = МО + 2 Nа (при избытке M)
С металлическим натрием расплав NаОН реагирует по схеме:
2 Nа + NаОН Ы NaH + Na2O
Нагревание гидроксида лития выше точки плавления ведет к термическому разложению по схеме
2 LiOН = Li2O + Н2О
которое становится заметным примерно важных для техники веществ — едкой щелочи и свободного хлора. В качестве побочного продукта получается также водород.
Важнейшим условием правильной работы электролитической установки является отсутствие взаимодействия между образующимися продуктами (щелочью и хлором), что может быть достигнуто возможно полным исключением перемешивания анодной и катодной жидкостей. При особенно часто применяемом диафрагменном методе (рис. Ч11-4) анодное и катодное пространства отделяются друг от друга диафрагмой из хорошо проницаемого для жидкостей асбестового картона. Анод изготовляется из графита, катод — из железа. В процессе злектролиза раствор щелочного хлорида непрерывно подается в анодное пространство, а из катодного непрерывно вытекает раствор смеси щелочного хлорида и щелочи. При его упаринании хлорид выкристаллизовывается (растворимость NаСl в 50 %-ном растворе NаOH равна лишь 0,9%). Полученный раствор NаОН выпаривают в железных чанах, после чего сухой остаток переплавляют.
Принципиальная схема установки для электролиза NаСl по ртутному методу показана на рис. Х111-23. В электролизере (А) с гра-
фитовым анодом (1) и ртутным катодом (2) натрий выделяется на ртути и образует амальгаму, которая переводится в разлагатель (Б), где разлагается горячей водой. Образующийся раствор NаОН идет на концентриронание, а ртуть перекачивается насосом (8) обратно в электролизер. Получаемая по ртутному методу шелочь отличается большой чистотой.
Ионы щелрчных металлов бесцветны. Почти все соли, образуемые ими с обычными кислотами, хорошо растворимы и противоположность выделяющимся, как правило, без кристаллизационной воды солям К, Rb и Сs для солей лития образование кристаллогидратов весьма характерно. Натрий занимает промежуточное положение. Соли щелочных металлов и слабых кислот вследствие гидролиза показывают в растворе щелочную реакцию. Комплексные соединения с ионом щелочного металла в качестве комплексообразователя малоустойчивы (они известны лишь для Li и Nа). Напротив, весьма многочисленны комплексные производные, у которых ионы щелочного металла располагаются во внешней сфере. Многие из подобных комплексов отличаются большой устойчивостью, которая по ряду Li-Сs обычно возрастает.
Из труднорастворимых солей натрия наиболее практически важен гексагидроксоантимонат — Nа[Sb(ОН)6], осаждением которого пользуются в аналитической химии для открытия натрия. У лития, как правило, малорастворимы соли слабых неорганических киcлот и хорошо растворимы — соли сильных. Для калия, рубидия, цезия и франция характерна малая растворимость перхлоратов и хлороплатинатов.
С солями щелочных металлов во многих отношениях сходны соли а м м о н и я. Этот комплексный катион с эффективным радиусом 143 пм по размерам и ряду свойств располагается между К и Rb: многие соли аммония кристаллизуются изоморфно с солями К и Rb, сходны с ними по растворимости и т. д.
Галогениды соли рассматриваемых элементов представляют собой довольно тугоплавкие кристаллические вещества, за исключением LiF (и отчасти NаF), хорошо растворимые в воде. Наибольшее практическое значение из них имеет NаСl. Помимо потребления с пищей (отсюда название — поваренная соль) громадные количества хлористого натрия используются промышленностью. Его ежегодное мировое потребление исчисляется десятками миллионов тонн.
Источниками промышленного получения NаCl служат, с одной стороны, природные залежи каменной соли, с другой — моря и соленые озера (Баскунчак и др.). Из залежей каменная соль просто выламывается и затем измельчается. Такая соль часто бывает настолько чиста, что не требует дальнейшей очистки. Из морей и соленых озер NаСl добывают упариванием раcсолов под действием солнца или вымораживанием воды. В настоящее время лишь изредка применяется обычный ранее способ выварки соли за счет сжигания топлива. Получаемая из рассолов соль часто бывает загрязнена примесями (главным образом ионов Са2+, Мg2+ и SO42- ) и во влажном воздухе отсыревает. Напротив, чистая поваренная соль негигроскопична. Из других галогенидов щелочных металлов громадное значение имеет КСl — основа калийных удобрений.
Большинство галоидных солей шелочных металлов кристаллизуется по типу NаС1. Исключениями являются СьС1, СвВг и Сз1, для которых характерна структура центрированного куба (рис. Х11-12).
Бесцветность и прозрачность кристаллов щелочных галогенидов обусловлены практическим отсутствием их взаимолействия с видимым светом. Как видно из рис. Х111-24, кристалл NaСl имеет участки поглощения лишь в ультрафиолетовой и инфракрасной областях. Уменьшение ионных радиусов благоприятствует повышению прозрачности в ультрафиолетовой, а их увеличение — в инфракрасной области. Поэтому лучше всего пропускает ультрафиолетовые лучи LiF (до l = 108 нм), а инфракрасные — СsI (до l = 54 мк). Так как в отношении прозрачности к тем и лругим лучам щелочные галогениды превосходят остальные обычные материалы (рис. ХII-58 и Х11-59), ими пользуются при конструировании некоторых оптических приборов. Активированные примесью солей таллия кристаллы NаI или СsI дают вспышки видимого света («сцинтилляции») под действием радиоактивного излучения, что также находит использование при конструкровании некоторых приборов.
Температуры (° С) и теплоты (кДж/моль) плавления щелочных галидов сопоставлены ниже:
| Li | Na | K | Rb | Cs | ||||||
| F | 848 | 27,1 | 995 | 32,6 | 856 | 28,4 | 798 | 25,9 | 682 | 21,7 |
| Cl | 607 | 20,1 | 800 | 28,0 | 772 | 26,3 | 717 | 23,8 | 645 | 20,1 |
| Br | 550 | 17,6 | 750 | 25,9 | 680 | 25,5 | 680 | 23,4 | 638 | 23,4 |
| I | 449 | 14,6 | 662 | 23,4 | 640 | 23,8 | 640 | 22,2 | 622 | 23,4 |
Сплав состава (мол. %) 46,5 LiF, 11,5 NаF и 42,0 КF плавится при 454 °С, т. е. гораздо ниже каждого из фторидов в отдельности. Закристаллизованные в трубке и медленно охлажденные нити СsВr и СsI имеют волокнистую текстуру и обладают высокой пластичностью.
Расплавы щелочных галидов имеют, в основном, ионный характер. Однако рентгеноструктурным исследованием было установлено, что среднее расстояние между катионами и анионами в жидкости несколько меньше (а между ионами одинакового заряда несколько больше), чем в кристалле. Например, для КСl вблизи температуры плавления оно равно 310 пм против 314 пм для кристалла. Так как это среднее расстояние имеет статистическую природу, в жидкости должны существовать и отдельные молекулы КСl (d = 267 пм).
Расплавы галидов Nа и его аналогов хорошо растворяют соответствующие свободные металлы, причем растворимость возрастает по рядам Nа °С для NаСI) наблюдается даже полная смешиваемость. Охлаждение расплава NаСl + Nа 700 сопровождается выделением кристаллов поваренной соли, окрашенных в синий цвет (который обусловлен, по-видимому, располагающимися в анионных вакансиях свободными электронами).
Интересны имеющие место в расплавах реакции взаимного вытеснения щелочных металлов, например, по схеме
КF + Nа Ы NаF + К
Равновесия этих процессов иногда оказываются смещенными в сторону вытеснения менее активным щелочным металлом более активного.
Из сплавов щелочных галидов друг с другом наиболее интересна эвтектическая система LiСl-КСl. Прежде всего ею обычно пользуются при электролитическом получении металлического лития. Затем она может служить средой для криоскопии и изучения некоторых реакций. Например, было показано, что растворенные в ней фторотитанаты Li, Nа и К диссоциированы по схеме М2ТiF6 = 2 М++ 2 F- + ТiF4, а для систем VO43-Ы VО3- + O- и 2 VO3-Ы V2О5 + O2- были найдены приближенные значения констант равновесия (соответственно 2·10-6 и 1·10-4).
При сплавлении каких-либо двух щелочных галидов происходит обмен ионами в соответствии со схемой АХ+ ВУ Ы АУ + ВХ. Направления смешений равновесия в подобных ионных системах без растворителя определяются тем, что энергетически выгоднее образование солевых пар из наименьших ионов, с одной стороны, и из наибольших — с другой. Например, система NаС1+ RbI более устойчива, чем система NаI+ RbCl. По-видимому, подобным же преобладанием взаимодействия одинаковых атомов над взаимодействием разных обусловлена структура металлических эвтектик.
При сравнительно низких температурах (вблизи точек плавления) пары щелочных галидов содержат не только простые молекулы ЭГ, но и некоторую долю полимеров (ЭГ)n. Устойчивость последних, в общем, уменьшается с ростом ионных радиусов, т. е. по рядам Li > Nа > К > Rb > Сs и F > Сl > Вr > I. Так, пар фтористого лития содержит приблизительно 49% LiF, 36 % Li2F2, 15% Li3F3 (и, возможно, очень иебольшие количества более высоких полимеров), пар хлористого натрия — 4 % NаCl, 25 % Nа2Сl2 и 1 % Nа3Сl3, а пар иодистого цезия — 97 % СsI и 3 % Сs2I2.
Структурно были изучены некоторые димеры Li2Г2. По-видимому, они представляют собой плоские ромбы. Для d(LiГ) и РГLiГ даются значения соответственно 223 пм и 108° (Сl), 235 пм и 110° (Вr), 254 пм и 116° (I).
Растворимость щелочных галогенидов показана дается в приводимой сводке:
Растворимость в воде (моль/л при 18 °С)
| Li | Na | K | Rb | Cs | |
| F | 0,1 | 1,1 | 15,9 | 12,5 | 24,2 |
| Cl | 18,6 | 5,8 | 4,5 | 7,2 | 10,9 |
| Br | 20,3 | 8,6 | 5,4 | 6,5 | 5,6 |
| I | 12,2 | 11,8 | 8,6 | 7,2 | 2,8 |
Минимум растворимости как будто намечается для солей элементов с близкими значениями атомных весов (NаF-КСl-RbВr-СsI). Однако подобные закономерности имеют случайный характер, так как с изменением температуры относительные растворимости отдельных солей могут меняться местами (например, NаСl и КВr). Для NаF было показано, что растворимость его до 100 °С возраcтает, а при дальнейшем повышении температуры последовательпо уменьшается.
Из растворов большинство щелочных галогенидов выделяется в безволном состоянии. Кристаллогидраты образуют перечисляемые ниже соли: LiСl·Н2О, LiВr·2Н2O, LiI·3Н2О, NаВr·2Н2О, NаI·2Н2О, КF·2Н2О, RbF·3/2Н2О, СsF·3/2Н2О. Ниже +0,15 °С в виде кристаллогидрата NаСl·2Н2О может быть выделен и хлористый натрий. Следует отметить, что кристаллы NаСl всегда содержат следы NаОН (наиболее чиста в этом отношении природная каменная соль).
Щелочные галогениды хорошо растворимы в жидком аммиаке. Из таких раствороы иногда могут быть выделены аммиакаты, например [Nа(NН3)4] (т. пл. 3 °С).
Растворимость щелочных галогенидов в органических растворителях, как правило, мала. Исключение представляют главным образом соли лития. Например, молярности их насыщенных растворов в безводном эфире при 25 'С составляют 3·10-4 (LiСl), 1,18 (LiВr), 3,48 (Lil), тогда как большинство друшх щелочных галидов в эфире практически нерастворимо. Относительно велика их растворимость в низших спиртах (г на 100 г растворителя при обычных температурах):
| NaCl | NaBr | NaI | KCl | KBr | KI | |
CH3OH | 1,4 | 17,4 | 77,7 | 0,5 | 2,0 | 16,5 |
C2H5OH | 0,07 | 2,3 | 43,1 | 0,003 | 0,14 | 1,75 |
На хлоридах было показано, что растворимость в спиртах меняется по ряду Li > Nа > К > Rb > Сs, увеличивается при нагревании и уменьшается с ростом молекулярного веса спирта (причем растворимость в изоспиртах выше, чем в нормальных). Интересен ход констант диссоциации хлоридов в смеси диоксана (70 %) с водой (30 %) — значения К·103 оказались равными: 2,9 (Li), 5,3 (Nа), 2,4 (К), 2,6 (Rb) и 2,7 (Сs). Для одновременно изучавшегося [N(СН3)4]Сl аналогичное значение равно 2,2. Таким образом, в этой смеси с e = 20 все рассматриваемые соли оказались довольно слабыми электролитами.
Отдельные щелочные галогениды находят разнообразное практическое использование. Так, NаF служит для пропитки древесины (железнодорожных шпал и т. д.) с целью предохрения ее от гниения. Бромистые и иодистые соли Nа и К применяют в медицине (часто под неправильными названиями «бром», «белый иод» и т. п.).
Интересными производными щелочных металлов (а также аммония) являются полигалогениды общей формулы МГn. Простейшим примером образований этого типа могут служить ионы I3-.
Полифториды не получены, а из полихлоридов были выделены лишь отдельные производные очень объемистых катионов (например, [N(СН3)4]Сl3). Напротив, полибромиды и полииодиды, а также многие смешанные полигалогениды типа МГ3, содержащие одновременно различные галоиды, изучены довольно хорошо. Для характеристики зависимости их устлйчивости от природы комплексообразователя ниже сопоставлены константы нестойкости ионов типа ГI2’, в водном растворе при 25 °С:
Г Cl Br I
[Г’]·[I2]/[ГI2-] = K K 6·10-1 8·102- 7·10-3
Безводные соли щелочных металлов типа ЭГ3 получены лишь для К, Rb и Сs (а также NН4). Цвет зтих соединений в зависимости от преобладания Сl, Вr или I изменяется от желтого через красный к почти черному. Зависимость их термической устойчивости от природы катиона и состава аниона видна из приводимых ниже данных по давлению диссоциации солей типа ЭIГ2 при 150 °С (мм рт. ст.):
RbIСl2 СsIСl2 RbIВr2 СsIВr2 RbI3 СsI3
507 114 248 39 30 12
По данным инфракрасной спектроскопии, ионы IСl2-, ВrСl2-, Вr3-,СlF2-, Сl3- линейны и симметричны. В первом из них d(IСI) = 255 пм.
По ряду Li-Сs растворимость полигалогенидов в воде уменьшается, устойчивость их возрастает. Наиболее устойчивым из полигалоидных ионов является [IСl4]-, желтые соли которого могут быть получены (действием хлора на концентрированные солянокислые растворы соответствующих иодидов) для всех щелочных металлов. В виде оранжево-желтого кристаллогидрата Н[IСI4)·4Н2О была выделена свободная кислота. По данным рентгеноструктурного изучения кристалла К[IСl4]·Н2О этот ион имеет форму искаженного квадрата с четырьмя разными ядерными расстояниями d(IС1) = 242, 247, 253 и 260 пм. По-видимому, такое их различие обусловлено не свойствами самого иона, а общей композицией кристалла.
При хранении на воздухе почти все полигалогениды постепенно распадаются с отщеплением свободного галогена. Быстрее этот распад протекает при нагревании или соприкосновении полигалогенида с хорошо растворяющими свободный галоид жидкостями (ССl4, эфир и т. п.). В случае смешанных полигалогенидов разложение происходит таким образом, что образуется соль щелочного металла с наиболее химически активным галоидом. Примером может служить термический распад по схеме
КIСl4® Сl2 + КIСl2® КCl + IСl
Гидриды лития и его аналогов (ЭН) образуются при пропускании сухого водорода над нагретым металлом. Взаимодействие сопровождается сильным уменьшением объема исходного металла {в %): 25 (Li), 26 (Nа), 40 (К), 41 (Rb), 45 (Сs). По внешнему виду и большинству физических свойств гидриды похожи на соответствуюшие галоидные соли. Так, лучше других изученный LiН образует твердые бесцветные кристаллы, в отсутствие воздуха плавящиеся без разложения при 668 °С. Солеобразная природа рассматриваемых гидридов была также непосредственно доказана выделением водорода при электролизе расплавленного LiН на аноде.
Взаимодействие водорода с нагретыми щелочными металлами идет медленнее, чем с щелочноземельными. В случае Li требуется нагревание до 700-800 °С, тогда как его аналоги взаимодействуют уже при 350-400 °С. Для достижения наибольшей полноты использования металла целесообразно вводить в реакционное пространство его пары. Можно также воспользоваться тем, что при нагревании в атмосфере водорода образующийся гидрид возгоняется (хотя и с трудом) и оседает затем на более холодных частях реакционного сосуда в виде бесцветных игол или похожий на вату спутанной кристаллической массы. Интересным способом образования рассматриваемых гидридов (наряду с окислами Э2О) является взаимодействие щелочного металла с его расплавленной гидроокисью.
Из индивидуальных молекул лучше других охарактеризован гидрид лития. Молекула LiН полярна (m = 5,88). Связь Li-Н имеет длину 160 пм и энергия диссоциации равна 234 кДж/моль. В молекуле гидрида натрия 189 пм.
По термической устойчивости гидриды Nа-Сs близки друг к другу — давление их диссоциации достигает 760 мм рт. ст. при следующих температурах (°С): 420 (Nа), 427 (К), 364 (Rb), 389 (Сs). В отсутствие избытка водорода все они разлагаются на элементы еще до достижения температур плавления. Напротив, LiН по устойчивости превосходит даже гидриды щелочноземельных металлов: давление его диссоциации при 500 °С составляет только 0,07 мм рт. ст. и достигает атмосферного лишь около 850 °С. Под давлением водорода температура плавления NаН лежит выше 800 °С (т. е. превышает температуру плавления NаСl).
Гидриды щелочных металлов являются очень сильными восстановителями. Окисление их кислородом воздуха в сухом состоянии идет сравнительно медленно, но в присутствии влаги процесс настолько ускоряется, что может привести к самовоспламенению гидрида. Особенно это относится к гидрилам К, Rb и Сs. С водой происходит бурная реакция по схеме ЭН + Н2О = Н2 + ЭОН или в ионах: Н-+ Н+ = Н2. При взаимодействии NаН или КН с двуокисью углерода образуется соответствующая соль муравьиной кислоты.
Заметно отличается LiН от своих аналогов также и по реакционной способности. При обычной темиературе в отсутствие влаги на него не действуют не только кислород воздуха, но и Сl2 и НСl. Напротив, при нагревании или в присутствии воды он ведет себя вполне аналогично гидридам других щелочных металлов. При хранении бесцветных кристаллов LiН на свету они постепенно окрашиваются в синий цвет. Появление этой окраски обусловлено, вероятно, частичным переходом элек.ронов с Н- в пустоты решетки.
Гидрид натрия используется иногда в металлургии для выделения редких металлов из их соединений. Его 2 %-ный раствор в расплавленном NаОН находит применение для снятия окалины со стальных изделий (после минутного выдерживания в нем горячее изделие погружают в воду, причем восстановившаяся по уравнению Fе3O4 + 4 NаН = 4 NаОН + 3 Fе окалина отпадает). При электролизе такого раствора на катоде выделяется натрий, а на аноде водород.
А з и д ы щелочных металлов могут быть получены по схеме
Э2СО3 + 2 НN3 = Н2О + CO2 + 2 ЭN3
Они представляют собой бесцветные кристаллические вещества, при нагревании начинающие разлагаться на элементы еще до достижения температур плавления. Растворимость их в обычных условиях составляет приблизительно 42 (Nа), 50 (К), 100 (Rb) и 300 (Сs) г на 100 г Н2О. Для энергий кристаллических рещеток даются значения (кДж/моль): 811 (Li), 732 (Nа), 656 (К), 635 (Rb) и 610 (Сs). С сероуглеродом в водной среде азиды реагируют по схеме КN3 + СS2 = КSCSN3, обрзуя азидодитиокарбонаты, а с иодом (в присутствии следов СS2) — по схеме 2 КN3 + I2 = 2 КI + 3 N2.
Н и т р а т ы щелочных металлов сравнительно легкоплавки и хорошо растваримы в воде. Практическое значение из них имеют почти исключительно NаNО3 и КNО3. Обе соли используются главным образом в качестве минеральных удобрений. Нитрат калия идет также для изготовления черного пороха (NаNО3 не применяется из-за его гигроскопичности).
Точки плавления н и т р а т о в щелочных металлов лежат не очень высоко (°С): 253 (Li), 306 (Nа), 334 (К), 310 (Rb), 406 (Сs). По мере повышения давления (до 10 тыс. ат) температура плавления LiNО3, NаNО3 и СsNО3 последовательно возрастает, тогда как у RbNО3 она проходит через минимум, а у КNО3 через максимум. Рентгеновское исследование расплавов NаNО3 и КNО3 поклзало, что их структура близка к кристаллической. Под вакуумом при 350-450 °С большинство рассматриваемых нитратов может быть отогнано из их расплавов без сушественного разъложения. Для температур такого разложения по схеме 2 ЭNО3 = 2 ЭNО2 + O2 даются значения от 475 (Li) до 585 °С (Сs). Молекулы LiNО3 и NаNО3 в парах имеет строение, аналогичное молекуле НNО3 с параметрами d(LiО) = 160, d(NaО) = 1,90, d(ОN) = 140, d(NО) = 122 пм, РЭОN = 105°, РО=Х=О = 134°.
Для сплава NаNО3 и КNО3 характерна эвтектика (при 55 мол. % KNО3) с температурой затвердевания 225,7 °С. Система эта обладает высоким значением криоскопической константы (20,75 град) и может быть использована как среда для криоскопии. Например, константы диссоциации PbСl2 и СsСl2, были в ней найдены равными соответственно 3·10-3 и 1·10-4.
При повышении температуры растворимость нитратов сильно увеличивается. Изломы на кривой для азотнокислого лития обусловлены различной растворимостью его отдельных гидратных форм. Так как азотнокислые соли других щелочных металлов кристаллогидратов не образуют, кривые их растворимости подобных изломов не имеют.
Для ннтратов К, Rb и Сs, помимо безводных солей, известны кристаллические продукты присоединения азотной кислоты общих формул ЭNО3·НNО3 и ЭNО3·2НNО3, вылеляющиеся из растворов соответствчющик нитратов, содержащих большой избыток свободной НNO3. Примерами могут служить СsNО2·НNО3 (т. пл. 104 °С с разл.) и СsNО3·2НNО3 (т. пл. 39 °С с разл.). Эвтектика системы НNО3-СsNО3 лежит при 13 мол. % соли и -62 °С.
Большая часть потребляемого NаNО3 получается в качестве продукта азотнокислотного производства (за счет поглощения щелочами оксидов азота из отходяших газов). Азотнокислый калий обычно получают обменным разложением КСl и NаNО3.
Н и т р и т ы щелочных металлов (ЭNО3) представляют собой бесцветные (или слегка желтоватые) кристаллические вещестна, очень легко растворимые в воде (LiNО3 — и в спирте). Их точки плавления (°С) равны: 220 (Li), 283 (Nа), 438 (K), 422 (Rb), 398 (Сs). Наиболее важны NаNО3 и КNО3. Используются они главным образом при органических синтезах.
Интересной реакцией образования нитритов щелочных (и щелочноземельных) металлов является взаимодействие их твердых гидроксидов с монооксидом азота по схеме:
2 ЭОН + 4 NО = 2 ЭNО2 + N2О + Н2О
Процесс медленно идет в обычных условиях, причем скорость его по ряду Li-Сs резко возрастает. При нагревании взаимодействие идет гораздо быстрее, но главным образом по другой схеме, а именно:
4 ЭOH + 6 NО = 4 ЭNО2 + N2 + 2 Н2О
Термический распад нитрита натрия протекает около 900 °С по суммарному уравнению:
4 NаNО2 = 2 Nа2О + 2 N2 + 3 O2
Под вакуумом при 350-500 °С нитриты натрия и калия могут быть отогнаны из их расплавов без существенного разложения.
Из п е р х л о р а т о в щелочных металлов хорошо растворимы в воле и некоторых органических растворителях только соли Li и Nа (г/100 г растворителя при 25 °С):
Н2О СН3ОН С2Н5ОН СН3СОСН3 СН3СООСН3 (С2Н5)2О
LiClO4 27,2 64,6 60,3 57,7 46,8 53,2
NаСlО4 67,7 33,9 12,8 34,1 8,8 0
КСlO4 1,3 0,1 0,01 0,15 0,001 0
В безводной хлорной кислоте при 0 °С их растворимость изменяется следующим образом (г/100 г): 0,106 (Li), 0,628 (Nа), 4,26 (К), 22,6 (Rb), 68,4 (Сs).
Термическая устойчивость перхлоратов растет по ряду Li-Cs, причем плавление сопровождается разложением. Исключением является LiСlO4, который плавится при 247 °С, а начинает разлагаться лишь при 500 °С. Соль эта (как и LiNО3), благодаря высокому весовому содержанию активного кислорода, представляет интерес для реактивиой техники. Образуемая обеими солями эвтектика (с 46,5 мол. % LiNО3) плавится при 172 °С.
П е р м а и г а н а т ы шелочных металлов по растворимости похожи на перхлораты. Их термическая устойчивость несколько ниже и для температур разложения даются значения (°С): 190 (Li), 170 (Nа), 240 (К), 259 (Rb), 320 (Сs).
Ввиду двухосновности у г о л ь н а я кислота образует со щелочными металлами соли двух типов — кислые (ЭНСО3) и средние (Э2СО3). Кислые карбонаты (бикарбонаты) характерны для всех щелочных металлов кроме Li. Из растворов они выделяются без кристаллизацианной воды в виде мелкокристаллических порошков. При обычной температуре бикарбонаты устойчивы, но при нагревании довольно легко переходят в соответствующие средние соли угольной кислоты:
2 ЭНСO3 = Э2СО3 + СО2 + Н2О
По ряду Nа-Cs термическая устойчивость бикарбонатов заметно возрастает. За исключением NаНСО3 рассматриваемые бикарбонаты хорошо растворимы. Вследствие гидролиза растворы их показывают очень слабощелочную реакцию. При нагревании этих растворов из них чястично выделяется СО2 (в соответствии с приведенным выше уравнением распада), и реакция становится сильношелочной. В соприкосновении с воздухом такое выделение СО2 растворами бикарбонатов очень медленно происходит и при обычной температуре. Практическое применение находит главным образом NаНСО3 («питьевая сода»), используемая в медицине, кондитерской промышленности и т. д.
Чистый бикарбонат натрия может быть получен действием СО2 на раствор соды по реакции, обратной процессу его термического распада. В насыщенный (при 30-40 °С) теплый раствор соды пропускают ток диоксида углерода. Через некоторое время начинается выпадение осадка NаНСО3.
Кристалл NfHCO3 слагается из цепей анионов НСO3-, cоединенных друг с другом посредством водородных связей (рис. Х111-26), в которых d(O-Н) = 107 и d(O...H) = 156 пм (с осцилляцией протонов между обоими положенлями), а катионы Nа+ располагаются в промежутках между такими цепями. Растворы этой соли характеризуются мало злвисящими от концентрации значениями рН» 8,4.
В кондитерском и булочном производствах NаНСО3 применяется в составе п е к а р с к и х п о р о ш к о в. Последние обычно представляют собой смесь бикарбоната натрия и кислой соли какой-либо другой, термически более устойчивой кислоты (например, NаH2РO4). С целью воспрепятствовать преждевременному взаимодействию обоих солей к смеси их часто добавляют крахмал. При замешивании пекарского порошка с тестом начинает идти реакция, например, по схеме
NаНСО3 + NаН2РO4 = Na2HPO4 + СО2 + Н2О
причем пузырьки выделяющегося диоксида углерода задерживаются в тесте. В процессе выпечки пузырьки эти от нагревания расширяются и сообщают тссту необходимую пористость.
Тонкий порошок NаНСО3 (с небольшими добавками веществ, предупреждающих его спекание) используется для с у х о г о о г н е т у ш е н и я. При выбросе из огнетушителя (под давлением СО2) он образует густое облако, хорошо защищающее человека от теплового излучения и тем самым дающее ему возможность подойти ближе к месту пожара. При разложении NаНСО3 под действием огня, помимо его подавления образующейся газовой смесью СО2+Н2О, на горящих материалах осаждается сухая пленка Nа2СО3, что также способствует их изолированию от воздуха. Подобнsе огнетушители с успехом применяются при тушении горящей нефти, масел и т. п.
Имеется указание на возможность получения бикарбоната лития смешиванием при 0 °С растворов LiСl и NН4НСО3. Последовательно промытый насыщенной СО2 холодной водой, спиртом и эфиром осадок имел состав Li2СО3·1,65Н2СО3 и выше 0 °С быстро разлагался. Более устойчивая форма LiНСО3 может быть, по-видимому, получена путем ослждения абсолютным спиртом насыщенного диоксидом углерода раствора Li2СО3, содержащего желатину. Сообщается, что такой (защищенный желатиной) LiНСО3, не разлагается и при обычных температурах.
Температура плавлепия к а р б о н а т о в щелочных металлов (в атмосфере СО2) лежит весьма высоко: Li2СО3 - 720, Nа2СО3- 854, К2СО3- 901, Rb2СО3- 873, Сs2СО3- 731 °С. Смесь 56 мол. % Nа2СО3+ 44 К2СО3 значительно более легкоплавка (т. пл. 710 °С), чсм каждая из этих солей в отдельности. При прокаливании карбонатов начинается их диссоциация на оксид металла и диоксид углерода. Термическая устойчивость карбонатов при переходе от Li к К возрастает, а затем от К к Сs вновь уменьшается.
Растворимость Li2СО3 составляет при обычных условиях около 0,17 моль/л и с повышением температуры уменьшается. Растворы Nа2СО3 показывают сильнощелочную реакцию (в 0,1 н. растворе рН = 10,9, а в 1 н. — 12,3). Смесь равных объемов 0,025 М растворов Nа2СО3 и NаНСО3 имеет рН = 10,02 (при 25 °С).
При взаимодействии с пероксидом водорода карбонаты щелочных металлов способны образовывать пероксосольваты Э2СО3·nН2О2, причем склонность к этому растет от лития (для которого вопрос об их существовании пока не ясен) к цезию. Для натрия характерно n = 1 или 2, для К, Rb, Сs — 3 (но для Rb и Сs возможны и более высокие значения n). Карбонат цезия отличается способностью обрзовывать кристаллосольват с метиловым спиртом — Сs2СО3·6СН3ОН.
Помимо непосредственного использования соды при стирке белья на ее основе иногда готовят специальные составы для этой цели. Подобпий с т и р а л ь и ы й п о р о ш о к может содержать, например, 88 % соды, 10 % гипосульфита и 2 % буры.
Нормальные карбонаты щелочных металлов, за исключением Li2CO3, хорошо растворимы в воде, причем в результате гидролиза растворы их показывают сильнощелочную реакцию. Наибольшее значение имеет сода (Na2CO3). Вырабатывается она или в безводном состоянии (“кальценированная сода”) или в виде выветривающегося на воздухе кристаллогидрата Na2CO3·10H2O (“кристаллическая сода”).
Потребителями соды являются многие отрасли промышленности. Кроме того, она применяется для умягчения воды и стирке белья и т. д. Выработка кальцинированной соды по СССР составила в 1972 г. 3850 тыс. т (против 536 тыс. т 1940 г. и 160 тыс. т в 1913 г.).
Основное значение для производства соды имеет аммиачный метод, основанный на реакции
NаСl + NН4НСО3Ы NаНСО3Ї + NН4Сl
равновесие которой почти нацело смещено вправо (вследствии очень малой растворимости NаНСО3 в растворе NН4Сl). Концентрированный раствор NaCl cперва насыщают аммиаком, а затем обрабатывают диоксидом углерода (получаемой за счет обжига СаСО3). Выделяющийся NаНСО3 отфильтровывают и нагреванием переводят в Nа2СО3 (по приведенному выше уравнению), причем образующийся СO2 возвращают в производство. Содержащий NН4Сl маточный раствор обрабатывают гашеной известью и выделяющийся при этом аммиак также возвращают в производство. Таким образом, елинственным отходом является остающийся в растворе СаСl2.
Принципиальная схема заводской установки для получения соды по а м м и а ч н о м у методу (Сольвэ, 1863 г.) показана на рис. Х111-28. В печи (А) идет обжиг известняка, причем образующийся СО2 поступает в карбонизационную башню (Б), а СаО гасится водой (В), после чего Са(ОН)2 перекачивают в смеситель (Г), где он встречается с NН4Сl, при этом выделяется аммиак. Последний поступает в абсорбер (Д) и насыщает там крепкий раствор ХаС1, который затем перекачивают в карбонизационную башню, где при взаимодействии с СО2 образуются NaНСO3 и NН4Сl. Первая соль почти полностью осаждается и задерживается на вакуум-фильтре (Е), а вторую вновь перекачивают в смеситель (Г). Таким образом все время расходуются NаСl и известняк, а получаются NаНСО3 и СаСl2 (последний — в виде отхода производства). Бикарбонат натрия переводят затем нагреванием в соду.
При наличии природных источников Nа2SO4 рентабельным может быть и более старый, с у л ь ф а т н ы й способ производства соды (Леблан, 1791 г.). Последний осуществляется путем сплавления при 1000 °С смеси Nа2SO4, известняка и угля. При этом имеет место следующая реакция:
Nа2SO4 + 2 С + СаСО3 +217 кДж = 2 СО2 + Nа2СО3 + СаS
От труднорастворимого СаS сода отделяется путем обработки сплава водой. Отход производства — СаS — может служить исходным продуктом для получения из него Н2S и затем серы.
Природная сода содержится в воде содовых озер (Танатар и др.), которые имеются у нас в Западной Сибири. Образование в них соды обусловлено бактериальным восстановлением Nа2SО4 до Nа2S и переходом последнего в Na2СО3 под действием воды и углекислоты воздуха. Выделяющийся при этом сероводород связывается обычно имеющимися в воде соединениями железа, образуя черный ил FеS. Из таких озер сода может быть добыта испарением воды или ее вымораживанием.
Иногда на дне подобных озер накапливаются даже осадки соды в виде минерала т р о н ы — Nа2СО3·NаНСО3·2Н2О. Для кристаллов этого соединения характерно наличие ионов Н(СО3)23-, образоравшихся в результате объединения двух ионов СО32- короткой водородной связью [d(OO) = 253 пм]. Подобное же попарное объединение ионов СО32-, по уже двумя водородными связями [d(OO) = 252 пм], характерно для кристаллов КHCО3.
Кроме соды в значительных количествах потребляется промышленностью (преимущественно — стекольной) поташ — К2СО3. Помимо химических методов получения из природного КСl, лля выработки поташа широко используются отходы некоторых производств (зола растений и др.). 124) Выработкл поташа из природного КСl осуществляется двумя методами. Один из них сводится к обработке диоксидом углерода КОН, полученного электролизом раствора КСl. Другой основан на малой растворимости двойной соли КНСО3·МgСО3·4Н2O, образуюшейся при насыщении СО2 взвеси МgСО3·3Н2О в растворе КСl. Под действием МgО двойная соль разлагается затем на К2СO3 и МgСО3·3Н2О, который возвращается в производство.
Среднее содержание поташа в золе растений сопоставлено ниже (вес.%):
Береза Осина Сосна Крапива Картофельная ботва Полынь
20 23 27 30 60 75
Преимущественными источниками попутного получения поташа служат зола подсолнечника (содержит до 50% К2СО3), отходы сахарного производства и отходы от переработки овечьей шерсти.
Подобно карбонатам, сульфаты щелочных металлов тоже известны кислые (ЭНSO4) и средние (Э2SO4). В воде те и другие хорошо растворимы. Практическое значение имеют главным образом Nа2SO4 (в технике часто называемый просто сульфат) и К2SО4 особснно первый из них. Важнейшим их потребителем является стекольная промышленность. Кристаллогидрат Nа2SO4·10Н2О (“мирабилит” или “глауберова соль”) применяется в медицине кзк слабительное.
Промышленное получение Nа2SO4 и К2SО4 основано либо на их выделении из природных минералов, либо на обработке соответствующих хлоридов серной кислотой. В последнем случае сульфаты являются побочными продуктами производства соляной кислоты. Громадные количества мирабилита содержатся в воде Кара-Богаз-Гола.
Из растворов сульфаты К, Rb и Сs кристаллизуются в безводном состоянии, а соли Li и Nа выделяются при обычных условиях в виде кристаллогидратов Li2SO4·Н2О и Na2SO4·10Н2О. Первый из них весьма устойчив, а второй может существовать лишь ниже 32,4 °С. Температуры плавления безводных сульфатов лежат весьма высоко: Li2SO4- 860, Nа2SО4- 882, К2SO4 - 1069, Rb2SO4- 1066, Сs2SO4- 1019 °С. Летучесть их изменяется по ряду Li > Nа °С. Наименее летучий Nа2SO4 кипит при 1430 °С (по-видимому, с некоторым разложением).
В парах калий сульфат ион SO42- сохраняет обычное для него строение правильного тетраэдра [d(SO) = 147 пм], а ионы К+ располагаются на перпендикуляре к противоположным ребрам тетраэдра [РОKО = 59°, d(КО) = 245 пм].
Из б и с у л ь ф а т о в щелочных металлов наиболее важен NаНSO4. Он хорошо растворим в воде (г на 100 г Н2О): 40 при 0 °С и 100 при 100 °С. Его кристаллогидрат NаHSO4·H2O (т. пл. 58,5 °С), вероятно, представляет собой смешанную оксониевую соль: Na(ОН3)SO4. Безводный NаНSO4 плавится при 186 °С, а при дальнейшем нагревании с отщеплением воды переходит в пиросульфат
2 NаНSО4 = Н2О + Nа2S2O7
Известны и другие кислые сульфаты натрия, например Nа3Н(SO4)2 Имеются указаиие на то, что NаНSO4 (в количестве 7 кг/т) является хорошим консервантом для зеленой массы силоса.
С у л ь ф и т ы щелочных металлов (Э2SО3) представляют собой бесцветные кристаллические вещества, хорошо растворимые в воде. При обычных температурах соль натрия выделяется из растворов с 7Н2О (выше 33 °С — без воды), соль калия — с 2Н2О. Из б и с у л ь ф и т о в (ЭНSO3) соль натрия известна только в растворе (поступаюший в продажу препарат представляет собой смесь различных продуктов разложения и состоит главным образом из Nа2S2О5).
С у л ь ф и д ы щелочных металлов (Э2”S) представляют собой бесцветные твердые вещества, на воздухе постепенно разлагающиеся. Для теплот их образования из элементов даются следующие значения (кДж/моль): 447 (Li), 372 (Nа), 426 (К), 347 (Rb) и 339 (Сs). Они хорошо растворимы в воде и для них известны кристаллогидраты Э2S·nН2О, где n = 9 (Nа), 5 (К) или 4 (Rb, Сs). В результате гидролиза их растворы показывают сильнощелочную реакцию. Под действием кислорода воздуха постепенно идет окисление с образованием тиосульфата:
2 Nа2S + 2 O2 + Н2О = Nа2S2O5 + 2 NаОН
Аналогичные сульфидам с е л е н и д ы и т е л л у р и д ы известны для всех шелочных металлов, но гораздо хуже изучены. При обычных условиях они бесцветны (кроме желтоватого Сs2Те).
Практическое значение имеет почти исключительно сеннистный натрий. Получают его обычно путем восстановления Nа2SO4 c углем при 900 °C. Рсакция илет в основном по уравнению:
Nа2SO4 + 2 С + 222 кДж = 2 СО2 + Na2S
В присутствии соединений железа (играющих роль катализатора) Nа2SO4 может быть восстановлен до Nа2S водородом уже при 600 °С. В отсутствие воздуха он плавится при 1180 °С, а заметно испаряться начинает лишь выше 1300 °С. Сернистый натрий используется главным образом в производстве органических красителей и в кожевенной промышленности. Он является также одним из часто применяемых дегазаторов.
Кипячением раствора сульфида с избытком серы (или сплавление, сульфидов с серой) могут быть получены п о л и су л ь ф и д ы, из которых для К, Rb и Сs были выделены и изучены все члены ряда Э2Sn вплоть до n = 6, а для Na до n = 2. Гидролиз их растворов уменьшается по мере повышения n.
Из н и т р и д о в щелочных металлов (Э3N) легко образуется только Li3N (т. пл. 845 °С). Взаимодействие между литием и азотом медленно идет уже при обычных температурах и быстро при 250 °С.
Нитриды других щелочных металлов могут быть получены взаимодействием их паров с азотом в поле тихого электрического разряда (или термическим разложением соответствуюих азидов в вакууме). Все они более или менее сходны по свойствам с нитридом лития, но гораздо менее устойчивы. Так, Nа3N медленно разлагается на элементы уже при 200 °С, а нитриды К, Rb и Сs даже взрывчаты. Следует отметить, что имеющиеся данные по нитридам тяжелых щелочных металлов не очень надежны.
При нагревании в атмосфере водорода нитрид лития переходит в гидрид (с одновременным образованием аммиака). Наоборот, несколько более сильным нагреванием LiH в атмосфере азота может быть получен Li3N. В качестве промежуточных продуктов при том и другом направлении реакции образуются а м и д (LiNН2) и и м и д лития.
Для фосфидов щелочных металлов характерны формы Э3Р (где Э — Li, Nа, К) и Э3Р (где Э — Nа, К, Rb, Сs). Известен также фосфид лития состава LiP. Все эти вещества могут быть получены прямым взаимодействием элементов. Водой они легко разлагаются.
Из карбидов щелочных металлов (Э2С2) путем непосредственного взаимодействия элементов при нагревании образуются Li2C2 (теплота образования 59 кДж/моль) и отчасти Nа2С2. Последний и его аналоги могут быть получены взаимодействием щелочного металла с ацетиленом. При 50 (К) или 100 °С (Nа) образуются кислые ацетилиды (по схеме, например, 2 К+ 2 Н2С2 = 2 КНС2 + Н2), которые выше 200 °С распадаются на соответствующий карбид и ацетилен (2КНС2= К2С2 + Н2С2).
В чистом состоянии карбиды щелочных металлов представляют собой бесцветные кристаллические вещества. Все они (несколько менее других Li2С2) характеризуются своей исключительно высокой химической активностью. Даже в атмосфере таких газов, как SO2 и СО2, они самовоспламеняются. Взаимодействие их с водой сопровождается взрывом причем металл сгорает, а углерод выделяется в виде угля. Лишь при медленном доступе водяного пара разложение Li2C2 и его аналогов протекает сравнительно спокойно и сопровождается выделением ацетилена по схеме:
Э2С2 + 2 Н2О = 2 ЭОН + С2Н2
Силициды щелочных металлов могут быть получены из элементов при 600-700 °С (в замкнутой системе). Для лития известны сине-фиолетовый Li2Si (т. пл. 752 °C) и серебристо-серый Li4Si (т. пл. 633 °С с разл.), а для остальных щелочных металлов характерен тип ЭSi. Силициды этого типа чрезвычайно чувствительны к влаге, а взаимодействие их с водой имеет взрывной характер. Выше 350 °С в вакууме NaSi распадается на элементы, тогда как производные калия, рубидия и цезия с отщеплением большей части металла переходят в силициды ЭSi8.
Из б о р и д о в щелочных металлов описаны NаВ6 и КВ6. Оба они могут быть получены взаимодействием элементов при температурах порядка 1000 °С (под давлением). По отношению к нагреванию и воде они устойчивы. Термическая диссоциация КВ6 в вакууме (10-4 мм рт. ст.) начинается лишь при 750 °С.
Из сопоставления свойств щелочных металлов и их соединений видно, что по ряду Li-Сs они в общем изменяются весьма закономерно. Подобно тому как это имело место у В и Ве, первый элемент подгруппы — литий — занимает несколько особое положение. Малая растворимость его солей с анионами СО32-, РО43- и F-, а также гидроксида, сравнительная легкость отщепления от нее воды при нагревании и некоторые другие свойства приближают литий к магнию и кальцию. Однако в основном литий все же является типичным щелочным металлом.