Реферат: Адсорбция полимеров на границе раздела твердое тело - водный раствор
СОДЕРЖАНИЕ
| Введение | ..................... 2 |
| Адсорбционное взаимодействие на границе раздела фаз и свойства граничных слоев | .................... .4 |
Граничные слои полимеров на твердых поверхностях | .................... .5 |
Влияние адсорбционного взаимодействия на молекулярную подвижность полимерных цепей в граничных слоях | .................... .6 |
| Изменение свойств граничных слоев как следствие уменьшения молекулярной подвижности | .................... .9 |
| Влияние твердой поверхности на надмолекулярные структуры полимеров | ................... .13 |
О связи адсорбции полимеров с адгезией полимеров к поверхностям | ................... .13 |
| Влияние границы раздела на реакции синтеза и структуру трехмерных полимеров | ................... .15 |
| Заключение | ................... .21 |
| Литература | ................... .23 |
ВВЕДЕНИЕ
Одним из важнейших разделов физической химии полимеров и коллоидной.химии в настоящее время является физико-химия поверхностных явлений в полимерах [1,2]. Это связано с тем, что создание новых полимерных материалов, начиная от применяющихся в бытовых целях и кончая космической техникой, непосредственно связано с использованием гетерогенных полимерных систем Действительно, большая часть современных полимерных материалов является гетерогенными системами с высокоразвитыми поверхностями раздела фаз. Это — армированные пластики, наполненные термопласты, усиленные резины, лакокрасочные покрытия, клеи и др.
Вследствие этого поверхностные явления в полимерах и полимерных материалах играют существенную роль во всем комплексе их свойств, и прежде всего, в структурно-механических свойствах, а исследование особенностей поведения макромолекул на границе раздела фаз является сейчас одной из важнейших задач в этой области. Говоря о проблеме поверхностных явлений в полимерах, нельзя забывать, что она имеет важное значение не только с технической точки зрения, но и с биологической, поскольку роль поверхностных явлении в биологических процессах, где принимают участие молекулы биополимеров, также очень велика. Наконец, проблема существенна и для решения вопросов новой развивающейся области — применения полимеров в медицине, где поверхностные явления происходят на границе раздела фаз с живыми тканями.
Проблема адсорбции полимеров — весьма разносторонняя и многообразная. Она включает такие важные для техники вопросы, как адгезию полимеров к твердым поверхностям, структуру и свойства монослоев, структурно-механические свойства граничных слоев полимеров, находящихся в контакте с твердыми телами, и многие другие. Однако все эти вопросы тесно связаны с одним, центральным, вопросом всей проблемы — адсорбцией полимеров на твердых поверхностях.
Действительно, адгезионное взаимодействие на границе раздела полимер — твердое тело есть, прежде всего, адсорбционное взаимодействие между двумя телами. Адсорбция полимеров на поверхности твердого тела определяет особенности структуры граничного слоя, характер упаковки макромолекул в граничных слоях, а отcюда — молекулярную подвижность цепей и их релаксационные и другие свойства. Процессы адсорбции играют существенную роль не только в комплексе конечных физико-химических и физико-механических свойств полимерных материалов, но и в ходе формирования полимерного материала, при его переработке пли синтезе в тех случаях, когда эти процессы протекают в присутствии твердых телиной природы — наполнителей. пигментов, на поверхности металлов, стекла и др. Образование клеевых соединений, нанесение лакокрасочных покрытий и ряд других технологических процессов включают в себя как первую стадию адсорбцию полимеров из поверхностности. Отсюда вытекает важная роль исследования процессов адсорбции полимеров на твердых поверхностях в большинстве технологических процессов.
Несмотря на то, что процессам адгезии в мировой научной литературе посвящено очень большое число работ [3-14], истинный механизм адгезии с молекулярной точки зрения изучен еще недостаточно. Существующие и развивающиеся теории адгезии носят частный и ограниченный характер. Электрическая теория адгезии [3-4] рассматривает электрические явления, возникающие при отслаивании адгезии от подложки, но не объясняет и не может объяснить самой адгезии, ибо электрические явления возникают в процессе расслоения, а адгезия нас интересует в условиях, когда адгезионная связь не нарушена. Диффузионная теория адгезии [14] применима практически только для случая адгезии полимеров друг к другу. Таким образом, единственно приемлемой сегодня будет адсорбционная теория адгезии, связывающая адгезию с действием межмолекулярных сил на границе раздела, т. е. с адсорбцией. Обладая рядом ограничений, присущих любой теории, с физической точки зрения адсорбционная теория является наиболее обоснованной. В частности, представления о возникновении двойного электрического слоя при контакте разнородных поверхностей также есть результат адсорбции и ориентации полярных групп макромолекул на поверхности, т. е. эти представления укладываются в рамки адсорб-цинной теории [4]. Однако развитие этой теории тормозится из-за недостаточной разработанности теории адсорбции макромолекул на твердых поверхностях.
Обобщение и развитие представлений об адсорбции должно стать фундаментом для дальнейшего развития физической химии наполненных и армированных полимеров, а также физико-химии нетканых полимерных материалов, играющих важную роль в современной промышленности.
АДСОРБЦИОННОЕ ВЗАИМОДЕЙСТВИЕ НА ГРАНИЦЕ РАЗДЕЛА ФАЗ И СВОЙСТВА ГРАНИЧНЫХ СЛОЕВ
Адсорбция полимеров на границе раздела фаз с твердым телом играет важную роль в усиливающем действии наполнителей, адгезии, склеивании и т. п. Адсорбционное взаимодействие является одним из важнейших факторов, определяющих свойства наполненных и армированных полимеров, свойства клеевых прослоек, адгезию полимеров и др. Совершенно очевидно, что многие особенности структуры адсорбционных слоев, получаемых при адсорбции полимеров на твердой поверхности из жидкой фазы, должны сохраниться и в таких системах, в которых адсорбционное взаимодействие полимера с твердой поверхностью реализуется в отсутствие растворителя, т. с. во всех практически важных системах (армированных и наполненных пластиках, покрытиях, клеях и т. п.). Для понимания свойств систем и нахождения путей их регулирования крайне важно знать структуру адсорбционных слоев в таких гетерогенных полимерных материалах. Между тем адсорбционные методы, позволяя выявить ряд существенных черт взаимодействия полимеров с твердыми поверхностями и поведения полимеров на границе раздела, не могут дать полных сведений о структуре граничных слоев в полимерных материалах. Это связано с тем, что адсорбционные взаимодействия в растворе не идентичны таковым в отсутствие растворителя. Последнее обстоятельство обсловлено отличием конформаций макромолекулярных цепей в растворе от конформаций в высокоэластическом, стеклообразном или кристаллическом и вязкотекучем состояниях.
ГРАНИЧНЫЕ СЛОИ ПОЛИМЕРОВ
НА ТВЕРДЫХ ПОВЕРХНОСТЯХ
Молекулярная подвижность полимеров в граничных слоях определяется гибкостью полимерной цепи н характером ее взаимодействия с поверхностью, т. е. теми же факторами, которыми определяется адсорбция. При рассмотрении вопроса о молекулярной подвижности следует иметь в виду, что прямое определение молекулярной подвижности в адсорбционных слоях полимеров экспериментально затруднено и лосих пор в литературе отсутствуют работы, в которых такие исследования были бы проведены действительно на адсорбционных слоях.
Мы имеем в виду необходимость разграничения понятий об адсорбционном и о граничной слое. В соответствии с изложенным, адсорбционным слоем является тот слой макромолекул, который образуется на поверхности вследствие адсорбции на ней полимера из раствора и в котором часть сегментов полимерных цепей находится во взаимодействии с поверхностью Толщина такого адсорбционного слоя определяется конформацией адсорбированных молекул, но уже при переходе к более сложным системам, в которых имеет место полимолекулярная адсорбция или адсорбция на поверхности не отдельных макромолекул, а их агрегатов, такое определение становится уже не применимым, так как в этом случае с поверхностью оказываются связанными не только молекулы полимера, имеющие непосредственные контакты с поверхностью. На такую возможность указано в работах Силберберга, а также в работах Ю С. Липатова н Л. М. Сергеевой [15-17].
Условия образования подобных систем исключают также возможность непосредственного исследования свойств граничных слоев Практически нигде (за исключением кристаллизующихся в очень тонких слоях полимеров) нельзя исследовать свойства собственно граничных слоев, и поэтому все выводы делаются на основании изменений, вносимых границей раздела в объемные свойства полимера, т. е. на нахождении некоторых избыточных характеристик. Поэтому все экспериментальные характеристики являются суммой свойств граничного слоя и объема, и суждения о характере изменения структуры в граничных слоях делаются на основе анализа направления изменения тех или иных характеристик. В этом случаенаиболее удобней моделью для исследования свойств граничных слоев являются наполненные полимеры, которые можно рассматривать как систему из частиц твердого тела с тонкими полимерными слоями на поверхности.
ВЛИЯНИЕ АДСОРБЦИОННОГО ВЗАИМОДЕЙСТВИЯ НА МОЛЕКУЛЯРНУЮ ПОДВИЖНОСТЬ
ПОЛИМЕРНЫХ ЦЕПЕЙ В ГРАНИЧНЫХ СЛОЯХ
Адсорбционное взаимодействие полимерных молекул с поверхностью, которое имеет место в наполненных системах, можно рассматривать как процесс, приводящий к перераспределению межмолекулярных связей в системе и к образованию дополнительных узлов физической структурной сетки вследствие взаимодействия сегментов с поверхностью. Образование дополнительных узлов должно снижать молекулярную подвижность как результат структурирования системы. Можно ожидать, что в зависимости от условий получения наполненного полимера и типа взаимодействия цепей с поверхностью число дополнительных узлов будет различно, а следовательно, и свойства поверхностного слоя полимера также будут отличаться. Первым актом образования поверхности и пленки (лакового, покрытия, клеевого соединения и т. п.) является адсорбция молекул полимера поверхностью. В зависимости от характера адсорбции и формы цепей в расплаве или растворе свойства поверхностных слоев будут различными.
Исследование релаксационных процессов в полимерах, находящихся на границе раздела с твердыми телами, представляет теоретический и практический интерес в связи с проблемой создания конструкционных наполненных полимерных материалов и нахождения оптимальных условий переработки и эксплуатации.
Установлено [18], что наличие границы раздела приводит к существенному изменению релаксационного поведения полимера в граничном слое, изменению температур стеклования н ширины интервала стеклования. изменению средних времен релаксации и пр. Это связано с изменениями плотности молекулярной упаковки, а также с уменьшением подвижности сегментов полимерных цепей и более крупных кинетических элементов вследствие их взаимодействия с твердой поверхностью.
На основании данных авторы считают, что ограничения подвижности целей в граничных слоях связаны прежде всего с энтропийным фактором, т. е обеднением конформационного набора макромолекул вблизи границы раздела. Эго позволяет удовлетворительно объяснить независимость аффекта от химической природы поверхности, распространение изменения подвижности на слои,непосредственно не контактирующие с поверхностью. влияние на эти эффекты гибкости полимерной цепи. Действительно, конформационный набор молекул жесткоцепного полимера, который весьма ограничен по сравнению с гибкими молекулами, не может столь же сильно изменяться вблизи границы раздела вследствие жесткости цепей, как в случае гибких молекул. Здесь эффекты изменения подвижности цепей не проявляются.
Таким образом, можно заключить, что изменения молекулярной подвижности связаны с уменьшением гибкости цепи в граничном слое вследствие конформационных ограничений, накладываемых геометрией поверхности. При этом не имеет значения, вызвано ли изменение конформаций только наличием поверхности или некоторой степенью связывания молекул поверхностью Последний фактор, весьма существенный с точки зрения прочности адгезионной связи, не имеет существенного значения при уменьшении молекулярной подвижности, поскольку эти процессы не связаны с нарушением связей на границе раздела.
Следует отметить, что во всех приведенных примерах не рассматривались случаи сильных специфических взаимодействий на границе раздела, где, возможно, картина будет несколько отличаться от описанной.
С изложенной точки зрения представляется интересным оценить вклад энергетического и энтропийного фактора в изменение молекулярной подвижности вблизи границы раздела [35]. Это сделано на основании данных по энергиям активации релаксационных процессов в поверхностных слоях, полученных температурной зависимости средних времен релаксации (табл. 1).
где F — с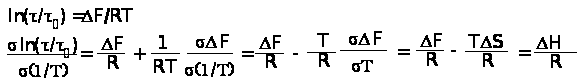
![]()
вободная энергия активации релаксационного процесса;
— время релаксации процессов;
0 — значение при 1/Т=0.
Из этого уравнения имеем:
или
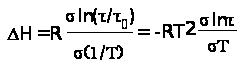
где H — энтальпия активации при условии независимости 0от Т. Отсюда
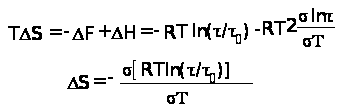
Таблица 1.
Значение активации и температурные смещения релаксационных процессов полимеров, находящихся в тонких слоях, определенных методом ЯМР и диэлектрическим методом
| Содержание аэросила, % | Содержание фторпласта, % | Энергия активации релаксации, ккал/моль | Энергия активации диэлектрической релаксации, ккал/моль | ||||
| ПММА | ПСТ | Сополимер ММА-СТ | ПММА | ПСТ | Сополимер ММА-СТ | ||
| Групповое движение | |||||||
| 1,8 | - | 2,1 | 23,7 | - | 14,9 | ||
| 8,83 | - | - | - | 1,7 | - | - | 12,6 |
| 1,32 | - | 1,4 | - | - | 18,5 | - | - |
| 23,08 | - | 1,2 | - | - | 15,4 | - | - |
| 24,90 | - | - | - | 1,8 | - | - | 10,7 |
| - | 26,5 | - | - | 1,7 | - | - | 12,6 |
| - | 49,2 | 1,5 | - | - | 20,0 | - | - |
| - | 75,0 | 1,4 | - | 1,5 | 18,8 | - | 10,6 |
| Сегментальное движение | |||||||
| 14,5 | 11,3 | 13,3 | - | 90,0 | 99,0 | ||
| 8,83 | - | - | - | 12,0 | - | - | - |
| 1,32 | - | 9,8 | - | - | - | 60,9 | - |
| 23,08 | - | 9,2 | 12,3 | - | - | 57,1 | - |
| 24,90 | - | - | - | 11,5 | - | - | 85,5 |
| - | 26,5 | - | - | 12,0 | - | - | 89,5 |
| - | 49,2 | 11,0 | - | - | - | 69,2 | - |
| - | 75,0 | 10,1 | 13,1 | 11,4 | - | 63,2 | 84,6 |
Таким образом, экспериментальные зависимости дают возможность определить термодинамические характеристики активационного процесса.
В поверхностных слоях по сравнению с объемом наблюдается заметное увеличение изменения энтропии активации в то время как энтальпия весьма незначительно уменьшается Эти результаты также показывают, что в изменение молекулярной подвижности цепей вблизи межфазовой границы основной вклад вносят конформационные эффекты.
Следует обратить внимание еще на одно обстоятельство. Изменение молекулярной подвижности в граничных условиях нельзя рассматривать как следствие адсорбционного взаимодействия, обусловленного только изменением теплосодержанием системы. В принципе одинаковые результаты можно получить для систем с сильным и слабым взаимодействием цепей с поверхностью, где все эффекты изменения молекулярной подвижности обусловлены энтропийными факторами. Соответственно, изменения подвижности не могут служить также характеристикой адгезии полимера к поверхности. Последующие исследования молекулярной подвижности в наполненных системах подтвердили основные положения, развитые в работах [22, 23].
ИЗМЕНЕНИЕ СВОЙСТВ ГРАНИЧНЫХ СЛОЕВ КАК СЛЕДСТВИЕ УМЕНЬШЕНИЯ МОЛЕКУЛЯРНОЙ ПОДВИЖНОСТИ
Ограничение молекулярной подвижности вследствие адсорбционного взаимодействия ведет к существенным изменениям свойств поверхностных слоев полимеров. Они проявляются в плотности упаковки молекул в поверхностных слоях, в температурах стеклования и реликсационном поведении наполненных полимеров, а также в характере образующихся на поверхности надмолекулярных структур.
Плотности упаковки в граничных слоях
Исследования адсорбции паров полимерами позволяет рассчитать изменение термодинамических функций при сорбции. Расчет показывает, что это повышение не может быть обусловлено сорбцией паров на поверхности твердого тела, а вызвано только изменениями структуры. Если в качестве сорбента берется растворитель данного полимера, то можно рассчитать, пользуюсь обычными термодинамическими соотношениями, изменение парциальной свободной энергии при сорбции и при условии, что система является атермической, и изменение парциальной удельной энтропии полимера, находящегося в объеме и на поверхности.
Так, для сорбции паров этилбензола полистиролом, содержащим различное количество стекловолокна, найдено, что S2 повышается с увеличением содержания наполнителя в пленке полимера. В соответствии с классическими представления теории растворов, это означает, что молекулы полимера располагаются в наполненной системе большим числом способов, чем в объеме. Вместе с тем рост сорбции указывает на разрыхление молекулярной упаковки макромолекул в граничных слоях.
С точки зрения теории растворов важной характеристикой свойств полимеров является их набухание. В соответствии с теорией Флори, набухание определяется числом узлов в пространственной сетке полимера и может быть использовано для их определения.
При изучении зависимости степени набухания от содержания полистирола на поверхности стекловолокна установлено [20], что по мере увеличения толщины слоя полимера на волокне происходит закономерное снижение набухания, которое лишь при содержании полимера около 200% от веса волокна приближается к набуханию полимера в объеме. Эти данные не только подтверждают разрыхление упаковки молекул на поверхности, но и указывают на большое расстояние от поверхности, на котором еще сказывается ее влияние.
Рассматриваемый пример относился к отучаю отсутствия сильного взаимодействия полимера с поверхностью. Если таковое имеет место, картина может быть существенно иной. Как показали исследования зависимости эффективной плотности пространственной сетки полиуретанов трехмерной структуры, нанесенных на подложку, от толщины покрытия, возникают дополнительные связи с поверхностью, приводящие к увеличению плотности сетки. По мере увеличения толщины слоя, эффект падает и на расстоянии от поверхности 200 мкм становится неразличимым. Следовательно, влияние поверхности в случае полимера сказывается на большом от нее удалении.
Таким образом, термодинамические исследования указывают на значительные различия в структуре и свойствах поверхностных слоев. Аналогичные результаты были получены впоследствии во многих работах.
Эффекты разрыхления можно объяснить следующим образом. Возникновение адсорбционных связей с поверхностью в ходе формирования полимерного материала, способствуя дополнительному структурированию системы, заметно ограничивает подвижность полимерных цепей вблизи поверхности, что приводит к изменению условий протекания релаксационных процессов и замедлению установления равновесного состояния полимера вблизи поверхности, а следовательно делает невозможным появление плотноупакованной структуры в таких условиях. Влияние условий протекания релаксационных процессов на плотность упаковки полимеров показано в работе [21].
Одновременно, что на поверхности происходит частично и сам процесс формирования надмолекулярных структур.
Можно допустить, что по тем же причинам агрегаты молекул или другие надмолекулярные структуры будут менее плотноупакованными. Чем больше поверхность наполнителя, тем больше ограничивается подвижность цепей уже в ходе формирования поверхностного слоя, и тем рыхлее упаковка в нем макромолекул. Посте завершения процесса формировании материала, когда агрегаты и молекулы более рыхлоупакованные, связаны с поверхностью, основное влияние на свойства имеет уже ограничение подвижности молекул. входящих в поверхностный слой.
Температуры стеклования граничных слоев
Как известно, переход из высокоэластического в стеклообразное состояние является кооперативным процессом, и поэтому величина скачка теплоемкости при стекловании зависит, очевидно, от числа молекул или сегментов, принимающих участие в переходе. Так как стеклование связано с проявлением подвижности макромолекул, то понижение скачка теплоемкости при стекловании может быть однозначно связано с исключением некоторой части макромолекул из участия в процессе. Экспериментальные данные подтверждают это положение: во всех случаях с ростом содержания твердой фазы скачок теплоемкости уменьшается. Это дает возможность подойти к оценке доли полимера, находящегося в граничных слоях. Если предположить, что макромолекулы, находящиеся в граничных слоях вблизи поверхности, не участвуют в общем процессе, то доля «исключенных» макромолекул составляет
= (1-f) = 1 — C/Ca,
где Ca, C — значение скачка теплоемкости для ненаполненного и наполненного образцов соответственно. Отсюда можно определить толщину граничного слоя следующим образом. Если упрощенно представить частицы наполнителя в виде сфер радиуса r, a толщину адсорбционного слоя обозначить через r, то объем адсорбционного слоя вокруг частички наполнителя будет описываться уравнением:
V = 4[(2+r)3 — r3]/3
С другой стороны, объемную дано граничных макромолекул можно представить как (1-f)c, где f — доля несвязанных макромолекул; с — общая объемная доля полимера в системе. Приравнивая отношение объема адсорбированного слоя вокруг частицы к ее объему и отношение общей объемной доли граничных макромолекул к объемной доле наполнителя в системе, можно написать:
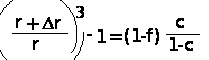
Если взять экспериментальное значение для системы олигоэтиленгликольадипинат — азросил (1-f) 1 и с = 0,975, то r/r 0,8. Так как частицы аэросила имеют диаметр около 250 А, то дм данной системы толщина слоя равна 100 А. Аналогичные величины порядка 170 А получены для наполненных сажей линейных полиуретанов.
Итак, абсолютное значение теплоемкости полимерной фазы в наполненных системах ниже, чем в ненаполненных, что интерпретируется как следствие понижения химического потенциала макромолекул в граничных областях по сравнению с химическим потенциалом в объеме. Таким образом, термодинамические данные указывают на определенные структурные изменения в граничных слоях полимеров на твердой поверхности.
Как уже было сказано — толщина граничного стоя зависят от свойств твердой поверхности и характеристик полимерной фазы. Влияние химической природы полимера на изменение свойств граничных слоев очень существенно. Рассмотрим некоторые литературные данные, полученные при измерении теплоемкости (табл. 2). Как видно из табл. 2 при увеличении в полимерах содержания аэросила во всех случаях происходит более или менее резкое понижение величины скачка теплоемкости Ср при температуре стеклования. Это указывает на переход некоторой части макромолекул из объема в граничные слои вблизи твердой поверхности. В табл. 2 приведены значения доли полимера в граничном слое, найденной из зависимости, учитывающей величину скачка теплоемкости при стекловании для наполненного и ненаполненного образцов. Значение увеличивается с повышением содержания наполнителя в системе (хотя пропорциональности при этом не наблюдается), и величина стремится к некоторому пределу.
Таблица 2.
Параметры стеклования в наполненных полимерах
Содержание аэросила, вес.% | Тс, С | Ср, кал/моль | | Ес, кал/моль | с, см3/моль | h, кал/моль | Vh, см3/моль |
Полистирол | |||||||
| 95 | 6,25 | - | 7320 | 100,5 | 1230 | 16,9 | |
1 | 95 | 5,60 | 0,105 | - | - | 1375 | 18,9 |
5 | 95 | 4,55 | 0,270 | - | - | 1705 | 23,5 |
10 | 95 | 3,10 | 0,505 | - | - | 160 | 29,7 |
15 | 95 | 3,00 | 0,520 | - | - | 2190 | 30,1 |
Полиметилметакрилат | |||||||
| 105 | 10,00 | - | 11380 | 85,9 | 1180 | 8,9 | |
1 | 110 | 9,80 | 0,020 | - | - | 1215 | 9,2 |
5 | 118 | 9,00 | 0,100 | - | - | 1350 | 10,2 |
7 | 121 | 8,40 | 0,160 | - | - | 1455 | 11,0 |
10 | 123 | 8,10 | 0,190 | - | - | 1530 | 11,5 |
Полиуретаны | |||||||
| -34 | 19,60 | - | 16380 | 143,0 | 895 | 7,8 | |
1 | -33 | 17,20 | 0,120 | - | - | 1020 | 8,9 |
5 | -32 | 15,80 | 0,195 | - | - | 1115 | 9,7 |
10 | -30 | 14,60 | 0,255 | - | - | 1170 | 10,2 |
20 | -30 | 14,20 | 0,275 | - | - | 1200 | 10,5 |
Полиметилсилоксан | |||||||
| -125 | 7,20 | - | 4985 | 65,0 | 695 | 9,0 | |
10 | -124 | 6,50 | 0,095 | - | - | 755 | 9,8 |
30 | -123 | 5,82 | 0,190 | - | - | 805 | 10,5 |
50 | -123 | 5,33 | 0,260 | - | - | 845 | 10,9 |
Vc — мольный объем полимера при Тс.
С точки зрения теоретических представлений об адсорбции интересно отметить результаты, полученные при исследовании температур стеклования пластифицированных наполненных полимеров [19, 22]. Найдено, что при одном и том же содержании пластификатора более резко снижается температура стеклования наполненного полимера по сравнению с ненаполнным. При повышении содержания пластификатора выше определенного предела температура стеклования наполненных пленок становится ниже, чем ненаполненных. Эти данные указывают на конкуренцию за места на поверхности между полимером и пластификатором, а также на вытеснение полимера с поверхности молекулами пластификатора, что соответствует представлениям об адсорбции смесей полимеров. Отметим также, что в работах Ю. С. Липатова и Т. Э. Геллер [18,19] на примере исследования объемной релаксации в на
24
полненных полимерах показано, что времена релаксации и энергии активации для данного типа полимера мало зависят от природы поверхности. Это подтверждает вывод о том, что в механизме ограничения подвижности цепей вблизи границы раздела наибольшую роль играют процессы, связанные с обеднением конфигурационного набора, которое определяется гибкостью макромолекул, а не энергетическим взаимодействием между полимером и поверхностью.
ВЛИЯНИЕ ТВЕРДОЙ ПОВЕРХНОСТИ НА НАДМОЛЕКУЛЯРНЫЕ СТРУКТУРЫ ПОЛИМЕРОВ
Адсорбционное взаимодействие на границе раздела фаз полимер- твердое тело, сказываясь на условиях формирования полимерного материала, приводит к изменению надмолекулярных структур граничных слоев и всей полимерной фазы в наполненной системе. В работе В. А. Каргина и Т. И. Соколовой [31] показано, что введение в кристаллизующиеся полимеры твердых добавок позволяет регулировать размер и число сферолитов. Механизм действия добавок заключается в том, что на поверхности твердых частиц в результате адсорбции возникают упорядоченные области полимера, играющие роль центров кристаллизации. С другой стороны, Ю. М. Малинским [32, 33] ингибирующее влияние твердой поверхности на кристаллизацию полимеров в пристенных слоях.
В работе [23] исследовано влияние твердой поверхности на над-молекулярные структуры в сшитых полимерах (полиуретанах), применяющихся в качестве покрытий на различных подложках. Найдено, что характер надмолекулярных структур определяется типом подложки и зависит от густоты пространственной сетки полимера. В этой работе впервые проведен послойный анализ надмолекулярных структур на разных удалениях от поверхности и показано, что по мepe удаления от нее характер морфологии изменяется и наблюдается переход от мелкоглобулярной плотноупакованной структуры к крупноглобулярной структуре с агрегацией глобул. Влияние поверхности на надмолекулярные структуры распространяется на большое удаление от поверхности. Лишь на удалении более 160 мкм структура пленок, сформированных на твердой поверхности, становится аналогичной структуре пленки, сформированной на границе раздела полимер — воздух.
О СВЯЗИ АДСОРБЦИИ ПОЛИМЕРОВ С АДГЕЗИЕЙ
ПОЛИМЕРОВ К ПОВЕРХНОСТЯМ
Адсорбционное взаимодействие на границе раздела фаз определяет адгезию полимеров к твердой поверхности. При этом следует говорить об адгезии в термодинамическом понимании как о работе, необходимой для преодоления сил сцепления двух различных поверхностей. С этой точки зрения должна существовать определенная связь между адсорбцией полимера на твердой поверхности и его адгезией. К сожалению, хотя во многих работах по исследованию адсорбции провозглашается задача установления связи адсорбции с адгезией, практически нигде такая задача не решалась, т. е. отсутствуют данные по сопоставлению адсорбции с термодинамической работой адгезии. Это может быть связано, прежде всего, с тем, что оценка термодинамической работы адгезии полимера к твердому телу экспериментально чрезвычайно затруднена. Вместе с тем обычно адгезию характеризуют не термодинамической равновесной величиной, а неравновесной работой обрыва.
Между тем отрыв, как указывает Я. О. Бикерман [12], практически никогда не происходит между двумя материалами. Всякое разрушение адгезионного соединения включает когезионное разрушение, и случаи истинно адгезионного разрушения редки, но даже и тогда отрыв является неравномерным. Поэтому прямое сопоставление найденных в различных работах характеристик адгезии с данными по адсорбции тех же полимеров к тем же поверхностям не может быть использовано для решения вопроса о связи адгезии и адсорбции. Поясним это положение на некоторых примерах.
Известно, что из трех полимеров — желатины, полистирола и полиметилметакрилата — наибольшую адгезионную прочность при неравновесном разрушении дает желатина. Адсорбция же желатины из растворов на поверхности стекла наименьшая. Фактически эти данные нельзя сравнивать, ибо адгезионная прочность обусловлена здесь другими причинам. Сопротивление разрыву в системе стекло — желатина — стекло превышает прочность склеек стекло — полистирол — стекло, во-первых, потому, что слабый граничный слой между влажной поверхностью стекла и желатиной (гидрофильным полимером) менее вероятен, чем между этой поверхностью и гидрофобным полимером; а во-вторых, потому, что когезионная прочность желатины обычно выше, чем полистирола, и при механическом нарушении склейки полистирола происходит когсзионный отрыв. Как видно из этого примера, ни тот, ни другой случай не имеет прямого отношения к адсорбции. Поэтому в принципе возможно только сравнение данных по адсорбции с термодинамически найденной работой адгезии. Но и здесь мы сталкиваемся со значительными трудностями, не позволяющими делать сравнение.
Действительно, как адсорбция, так н адгезия (в равновесном ее понимании) зависит от характера взаимодействия функциональных групп полимера с поверхностью, формы молекулы и пр. Однако условия возникновения адгезионной связи сильно отличаются от условий взаимодействия полимера и адсорбента в растворе. При адсорбции из растворов происходит конкуренция за места на поверхности между молекулами полимера н растворителя, которая снижает величину адсорбции полимера и прочность его связи с поверхностью.
Адсорбция сильно зависит от природы растворителя, поскольку последний определяет форму цепи, и. таким образом, условия контакта с поверхностью при адсорбции. При образовании адгезионной связи практически всегда, даже если нанесение склейки идет через стадию раствора, эти факторы исключаются полностью. При нанесении на поверхность растворов полимеров в растворителях, слабо взаимодействующих с поверхностью, адсорбция полимеров является первичным актом образования поверхностной или клеевой пленки.
Если раствори гель активно взаимодействует с поверхностью, то формирование адгезионного соединения начинается фактически только тогда, когда большая часть растворителя удалена из системы и возможно образование большого числа связей между полимерной молекулой и поверхностью в условиях, когда функциональные группы полимера уже не блокированы растворителем. В этом случае при удалении растворителя в ходе формирования пленки на поверхности происходит постепенное возрастание концентрации раствора и резко изменяется соотношение между сухарным числом взаимодействий полимерных молекул и молекул растворителя с поверхностью. Одновременно происходит и изменение структуры полимера, протекают процессы возникновения и релаксации внутренних напряжений, оказывающие влияние на прочность адгезионной связи [24, 25].
Следовательно, условия, при которых происходит адсорбция полимеров из растворов, и условия образования адгсзионной связи резко отличаются. Еще большим становится это различие, если адгезионное соединение получается не из раствора, а любым другим путем. Поэтому экспериментально нельзя установить прямой связи между адсорбцией полимера из раствора и адгезией его к данной поверхности, хотя она, безусловно, существует. Характер адсорбции определяет структуру возникающего на поверхности слоя, которая должна влиять на прочность адгезионной связи. В частности, это относится и к случаю определения адгезионной прочности неравновесными методами, ибо дефекты структуры слоев также связаны с условиями их формирования. При этом следует различать взаимосвязь адсорбции и адгезии (как равновесной характеристики, определяемой термодинамическими соотношениями), а также адсорбции и адгезионной прочности, понимая под последней неравновесную величину, определяемую путем нарушения адгезионной связи в неравновесных условиях.
ВЛИЯНИЕ ГРАНИЦЫ РАЗДЕЛА НА РЕАКЦИИ СИНТЕЗА И СТРУКТУРУ ТРЕХМЕРНЫХ ПОЛИМЕРОВ
В настоящее время благодаря работам школы В. А. Каргина установлено. что процессы возникновения структур в полимерах являются релаксационными процессами, зависящими от молекулярной подвижности структурных элементов цепей. Процессы структурообразования начинаются уже непосредственно в ходе полимеризации, и эти процессы взаимозависимы [26].
При создании полимерных материалов с заданным химическим строением и физической структурой особое значение имеет получение армированных пластиков н наполненных полимеров, в которых процессы полимеризации н одновременно структурообразования протекают в присутствии сильно развитой поверхности волокнистого или дисперсного наполнителя. Влияние малых количеств наполнителей, служащих центрами структурообразования в кристаллических полимерах, на процессы кристаллизации исследовано в работе [27].
Однако до сих пор еще мало исследованы процессы структурообразования при полимеризации в присутствии наполнителей, т. е. одновременное влияние поверхности раздела на протекание процессов полимеризации и структурообразования. Между тем эта проблема особенно важна при получении армированных и наполненных полимеров, где процессы полимеризации и структурообразования протекают на границе раздела с твердой поверхностью. В ряде исследований [23, 34] было изучено влияние твердой поверхности на процессы структурообразования при формировании полимерного материала из раствора или расплава и показано, что поверхность наполнителя оказывает существенное влияние на протекание этих процессов и свойства полимеров в граничных слоях.
Существенный интерес представляет также изучение влияния наполнителя на протекание реакции образования трехмерного полимера и его свойства.
Сильно развитая поверхность наполнителя на начальной стадии реакции может приводить к возрастанию скорости обрыва реакционных цепей на поверхности, в результате чего густота сетки уменьшается. и сетка становится более дефектной. Очевидно, поверхность наполнителя играет роль своеобразного ингибитора при формировании трехмерной сетки. Действительно, введение ингибитора при полимеризация в начале формирования сетки (при нарастании вязкости системы) привело к увеличению степени набухании полученного в отсутствие наполнителя трехмерного полимера при той же глубине превращения. Следовательно. ингибитор вследствие предотвращения реакции роста и сшивания также приводит к возникновению дефектной трехмерной структуры.
На глубоких стадиях реакции, вероятно, действует другой механизм, также приводящий к увеличению дефектности сетки. Из-за адсорбции растущих цепей полимера на поверхности наполнителя происходит значительное уменьшение их подвижности, также отражающееся как на скорости роста, так и на скорости обрыва. Все эти факторы способствуют возникновению более дефектной структуры трехмерной сетки.
Экспериментальное подтверждение влияния поверхности раздела на кинетику образования трехмерных полимеров можно показать на примере кинетики образования трехмерных полиуретанов в объеме и на поверхности [27]. Была изучена кинетика реакции образования полуретановых эластомеров путем сшивания триметилолпропаном макродиизоцианатов, полученных на основе полиоксипропиленгликолей с молекулярными весами 2000 и 1000, а также 4,4-дифенилметандиизоцианата при соотношении 1: 2. Кинетика образования полимера на медной подложке и в объеме исследовались методом ИК-спектроскопии.
Установлено, что скорость отверждения на подложке меньше для макродиизоцианата на основе полиэфира с меньшим молекулярным весом, в то время как в объеме для этого макродиизоцианата скорость выше. Поведение в объеме согласуется с тем, что уменьшение молекулярного веса полиэфира приводит к росту концентрации реакционноспособных изоцианатных групп в единице объема. Аномальное протекание реакции на поверхности может быть связано с тем, что увеличение концентрации сильно полярных групп NCО значительно увеличивает взаимодействие с подложкой исходного макродиизоцианата и образующегося полиуретана, которое существенно понижает подвижность цепей на поверхности для макродиизоцианата на основе полиэфира с меньшим молекулярным весом. Однако общая скорость реакции из поверхности в обоих случаях выше, чем в объеме. Это объясняется тем, что адсорбционное взаимодействие макродиизоцианатов с поверхностью приводит к определенной степени упорядоченности молекул друг относительно друга в поверхностном слое. Такая упорядоченность, согласно [28] способствует полимеризации и может приводить к возрастанию общей скорости процесса.
Таким образам, граница раздела оказывает двоякое влияние на процессы синтеза и структурообразования в трехмерных полимерах. увеличивая вероятность реакции обрыва на начальных стадиях реакции и затрудняя обрыв на более глубоких стадиях вследствие адсорбционного взаимодействия растущих цепей с поверхностью, которое, в свою очередь, влияет на скорость реакции и структуру сетки. В результате можно считать, что такая важная характеристика сетки, как эффективная плотность сшивки, учитывающая физические и химические узлы сетки, оказывается различной для случаев проведения реакции в присутствии и в отсутствие границы раздела с наполнителем. Это положение особенно хорошо иллюстрируется на примере изучения системы, в которой вклад физических узлов в эффективную густоту сетки очень велик по сравнению с вкладом химических узлов, а именно, на примере трехмерных полиуретанов [29]. В табл. 3 приведены найденные значения Мс — молекулярного веса отрезка цепей между эффективными узлами сетки.
Таблица 3.
Величины Мс и с/V для полиуретановых покрытий.
| Полиэфир | NCO: OH | Свободная пленка | Пленка на подложке | ||
Мс | с/V | Мс | с/V | ||
| Диэтиленгликольадипинат, мол. вес 800 | 2:1 | 620 | 21,0 | 210 | 61,0 |
| 1,75:1 | 970 | 13,4 | 130 | 100 | |
| 1,50:1 | 510 | 25,4 | 200 | 64,8 | |
| Поликсипропиленгликоль, мол. вес 2000 | 2:1 | 520 | 21,3 | 460 | 23,7 |
| 750 | 2:1 | 290 | 43,1 | 200 | 60,6 |
| 550 | 2:1 | 120 | 109 | 180 | 75 |
Как видно из таблицы, эффективная плотность сетки для пленок, находящихся на подложке, в большинстве случаев выше, чем для свободных пленок. С другой стороны, нет симбатности в изменении густоты химической сетки, задаваемой соотношением NСО/OН и физической густотой сетки (суммарной), что говорит о сложной взаимозависимости между химической структурой сетки, определяемой числом функциональных групп, участвующих в реакции, и строением сетки, обусловленные общим числом узлов. Прежде всего увеличение эффективной плотности сетки для пленок, находящихся на подложке, указывает на то, что адсорбция растущих цепей на поверхности в ходе реакции приводит к образованию дополнительных узлов сетки. Их число, в свою очередь, зависит от расстояния между химическими узлами сетки: чем оно больше, тем больше гибкость отрезка цепи между узлами и приспосабливаемость отрезков цепей к поверхности. При наибольшем Мс в свободных пленках наблюдается наименьшее Мс в пленке на поверхности (см. табл. 3).
Таким образом, различия в химической густоте сетки отражаются на свойствах пленок в свободном виде и на подложке. Отметим, что такие эффекты не могли наблюдаться для сополимеров стирола с дивинилбензолом, в которых отсутствуют функциональные группы, способные к сильному взаимодействию с поверхностью. В табл. 3 приведены также данные по влиянию молекулярного веса исходного полиэфира на эффективную плотность сшивки при одинаковом исходном соотношении NСО/ОН. С увеличением молекулярного веса полиэфира эффективная плотность сшивки уменьшается как длясвободных пленок, так и для пленок на подложке, что связано с уменьшением общей концентрации активно взаимодействующих с поверхностью функциональных групп.
Итак, наличие поверхности раздела вносит существенную специфику в формирование полимера трехмерной структуры, влияя на протекание химической реакции и на химическую и эффективную густоту трехмерной сетки, а отсюда — на структуру полимера, что особенно существенно с точки зрения регулирования свойств поверхностных пленок.
Дальнейшая задача в этой области заключается, по нашему мнению, в том, чтобы на основании кинетических и химических исследований установить детали механизма такого влияния и связать его с физической структурой образующегося на поверхности полимера. Это имеет большое значение для нахождения оптимальных путей получения армированных пластиков и наполненных полимеров, в которых, вследствие рассмотренных причин, происходят значительные изменения физико-химических свойств поверхностных слоев, а отсюда и физико-механических показателей. Решение указанной проблемы важно также с точки зрения установления общих связей между химическими и структурными превращениями в полимерах.
В заключение отметим, что в процессах отверждения на границе раздела существенную роль может играть различная адсорбируемость компонентов реакции. В тех случаях, когда отверждающаяся смола представляет собой смесь компонентов, проявляется избирательная сорбируемость какого-либо компонента поверхностью минерального наполнителя. Например. в системе эпоксидная смола — полиэтиленполиамин преимущественно сорбируется смола. Адсорбированная фаза не участвует в реакции отвержденпя. а смоляная фаза, обогащенная избыточным количеством отвердителя, становится менее жесткой, так как не вошедший в реакцию полиэтиленполиамин выполняет функцию пластификатора. Это понижает модуль упругости и увеличивает коэффициент термического расширения смоляной фазы. В системе метилололигометилфенолы — олигометиленфенолы в адсорбционном слое содержатся преимущественно высокомолекулярные компоненты, что сильно изменяет структуру отвержденной смоляной фазы.
Приведенные результаты позволяют сделать вывод о том, что на границе раздела с твердым телом, равно как н на границе раздела полимер — газ, происходит существенное уменьшение молекулярной подвижности полимерных цепей. Этот факт экспериментально доказан на большом числе аморфных полимеров с применением термодинамических, структурных и механических методов и считается сейчас твердо установленным. Однако вывод об изменении молекулярной подвижности явился результатом исследований свойств наполненных систем и покрытий, свойств, которые в конечном итоге опредялются молекулярной подвижностью.
Такие характеристики полимерного вещества, как температуры перехода из одного физического состояния в другое, вязкость, релаксационные характеристики и др., отражают молекулярную подвижность цепей и боковых групп. Поэтому все изменения данных характеристик, которые можно найти экспериментально, указывают на соответствующие изменения молекулярной подвижности.
Изменение молекулярной подвижности имеет следующие основные следствия. Оно ведет к повышению температур переходов, прежде всего — температуры стеклования, к изменению условий кристаллизации и к изменению релаксационного поведения полимера в поверхностных слоях. В последнем случае это влияние проявляется двояким образом: в ходе формирования полимерного материала из расплава или раствора, при полимеризации и в ходе эксплуатации уже готового полимерного материала. Ограничение молекулярной подвижности в поверхностных слоях при формировании полимера приводит к торможению релаксационных процессов и возникновению неравновесного напряженного состояния по сравнению с состоянием полимера в отсутствие твердой поверхности. В результате в системе возникает неплотная молекулярная упаковка и наполненный полимер может иметь в среднем меньшую плотность в расчете на полимер, чем ненаполненный.
С другой стороны, в случае трехмерных полимеров ограничение молекулярной подвижности в ходе синтеза сетки приводит к исключению части молекулярных цепей из участия в реакциях роста и формирования сетки, в результате чего возникающая сетка будет иметь большее число дефектов по сравнению с сеткой, полученной в присутствии поверхности раздела. Это — общая закономерность, установленная на большом числе систем.
Уменьшение плотности упаковки вместе с ограничением молекулярной подвижности изменяет условия протекания релаксационных процессов в уже сформированном полимерном материале, приводя к облегчению процессов, связанных с проявлением подвижности малых элементов цепей вследствие менее плотной упаковки, и к заторможению процессов, связанных с проявлением подвижности больших структурных элементов. При этом происходит расширение релаксационного спектра. Указанные следствия влияния поверхности наиболее существенные, хотя и сопровождаются изменением многих других характеристик полимерного материала.
Следует подчеркнуть, что мы говорим именно о свойствах полимерного материала, а не только о свойствах граничных слоев. В последнем случае мы не могли бы определить такие характеристики, которые проявляют только большое число молекул вместе и которые присущи собственно полимерному веществу. Это обстоятельство вместе с дополнительными экспериментальными данными дало нам основание считать, что действие поверхности распространяется на большом удалении от нее, т. е. влияние поверхности на подвижность непосредственно контактирующих с поверхностью цепей распространяется через другие цепи в объем материала. Дальнодействующее влияние поверхностных сил не есть, конечно, прямой результат действия силового поля поверхности, а является следствием общего изменения межмолекулярных взаимодействии в системе и изменения таких взаимодействий между цепями, непосредственно примыкающими к контактирующей с поверхностью цепи.
Это специфическая черта полимеров, а именно, сильные межмо-лекулярные взаимодействия между полимерными молекулами ведут к распространению эффекта влияния поверхности в объем. Фактически можно рассматривать участие молекулярных агрегатов или других надмолекулярных структур во взаимодействии с поверхностью. Ограничение подвижности хотя бы одной молекулы агрегата ведет к изменению поведения всех молекул данного агрегата.
И, наконец, что является причиной ограничения молекулярной подвижности цепей вблизи границы раздела? Самым простым ответом на данный вопрос было бы положение о чисто адсорбционном взаимодействии молекул с поверхностью. В подтверждение можно привести бесконечные данные о влиянии природы поверхности на адгезионные, механические и другие свойства поверхностных слоев полимеров. Действительно, адсорбционное взаимодействие — один из двух важнейших факторов, определяющих изменение молекулярной подвижности цепей вблизи границы раздела. Оно определяет, в частности, адгезию и прочностные свойства наполненных и армированных систем и клеев.
Но существует и другая причина изменения подвижности — чисто энтропийная, не связанная с энергетическим взаимодействием полимера с поверхностью. Вблизи границы раздела, как это следует из соответствующих расчетов, молекула не может иметь то же число конформаций, что и в объеме, поскольку поверхность накладывает ограничения на геометрию молекулы. В результате число состоянии молекулы в поверхностном слое уменьшается, уменьшается ее энтропия, что кинетически эквивалентно уменьшению молекулярной подвижности. Роль энтропийных факторов подтверждается расчетами энтальпии и энтропии активации при релаксационных процессах в поверхностных слоях. Теоретически возможны случаи равного изменения молекулярной подвижности в поверхностных слоях при разной величине энергии взаимодействия с поверхностью. При этом могут наблюдаться сопоставимые изменения характеристик, связанных с молекулярной подвижностью при резко различных механических свойствах, определяемых прочностью сцепления полимерной молекулы с поверхностью.
Таким образом, приведенные данные указывают на важную роль адсорбционного взаимодействия на границе раздела фаз полимер — твердое тело в процессах получения и использования гетерогенных полимерных материалов. Исследование адсорбции полимеров из растворов — средство выяснения закономерностей такого взаимодействия, которое существенно облегчает понимание процессов, происходящих в реальных системах, имеющих практическое значение.
ЗАКЛЮЧЕНИЕ
Анализ материала данного реферата, позволяет сделать некоторые общие выводы, касающиеся проблемы адсорбции полимеров на твердых поверхностях. Эти выводы базируются на современной теории разбавленных растворов полимеров и конфирмационной статистике полимерных цепей. Учет поведения макромолекул в разбавленных растворах, основанный на статистической термодинамике, позволил в настоящее время установить основные закономерности адсорбции полимеров и ее зависимость от природы полимера, поверхности, молекулярного веса и молекулярновесового распределения полимера, природы растворителя и температуры.
Результаты адсорбционных изменений позволили также сделать ряд существенных заключений о характере связывания полимерных молекул поверхностью адсорбента и их расположения на ней, а следовательно, и о структуре адсорбционного слоя. Теоретическое описание адсорбции в рамках конформационной статистики полимеров позволило в определенных пределах предсказать поведение полимерной молекулы в условиях адсорбционного взаимодействия с твердой поверхностью. Сложившиеся представления о механизме адсорбционных процессов имеют существенное значение для решения общих проблем поверхностных явлений в полимерах, в которых адсорбция играет доминирующую роль.
Вместе с тем совершенно очевидно, что теория адсорбции и адсорбционного взаимодействия полимерных молекул с поверхностями твердых тел еще разработана недостаточно для надежного предсказания адсорбции и объяснения многих экспериментальных фактов. В связи с этим остановимся на некоторых нерешенных проблемах. теории адсорбции. Ее построение должно основываться на уже установленных экспериментальных фактах, которых имеется достаточно много.
Действительно, в основу статистического рассмотрения кладутся представления о типах решеток, описывающих как раствор полимера, так и поверхность адсорбента, причем при выборе их параметров исходят из общих рассуждений, а не из представлений о конкретных системах. В зависимости от исходных предпосылок могут быть получены различные результаты. Их сопоставление с экспериментальными данными весьма затруднено, так как многие параметры, входящие в теоретические уравнения, не поддаются экспериментальному определению. Соответственно этому представления о структуре адсорбционного слоя также в известной мере зависят от модели и математических методов расчета.
Между тем вопрос о том, как меняется конформация цепи вблизи границы раздела, является центральным при рассмотрении структуры адсорбционных слоев и он не может быть решен только путем теоретических расчетов При обсуждении вопроса о конформациях в граничном слое следует иметь в виду, что ее изменения могут быть обусловлены как взаимодействием с поверхностью, сопровождающимся изменением энтальпии, так и энтропийным фактором, вследствие которого молекула вблизи границы раздела не может принять такого же числа конформаций, как в объеме.
При любом рассмотрения структуры адсорбционного слоя при полном заполнении поверхности адсорбента необходимо учитывать взаимодействие адсорбированных молекул друг с другом, которое также будет влиять на конформации молекулярных цепей в адсорбционном слое. Практически такое рассмотрение до сих пор не проводилось, хотя имеются указания на необходимость его учета.
Существующие представления о структуре адсорбционного слоя приводят еще к одной трудности. Она заключается в том, что адсорбционный слой рассматривается как раствор полимера. концентрация которого значительно выше концентрации полимера в фазе раствора. Тогда и термодинамическое, и статистическое описание поведения макромолекул должно отличаться от того, какое принимается для описания свойств разбавленного раствора. В этом случае нельзя пренебрегать возможностями агрегации макромолекул в адсорбционном слое. Введение представлений об агрегации макромолекул в адсорбционном слое, осложненном влиянием поверхности, является необходимым условием дальнейшего развития теории адсорбции.
Все трудности и проблемы, возникающие при рассмотрении теории адсорбции, относятся к теории адсорбции из разбавленных растворов. Переход к более концентрированным системам приводит к дополнительным осложнениям. В этом случае при изменении концентрации раствора происходят изменения как конформации макромолекул, так и условии их взаимодействия друг с другом. Возникновение молекулярных агрегатов, которое, как показано во многих работах, начинается уже в разбавленных растворах, приводит к тому, что при каждой концентрации раствора мы имеем дело с адсорбируемыми частицами, отличающимися друг от друга как по форме, так и по размеру. Соответственно этому изменяются условия контакта молекул и их агрегатов с поверхностью адсорбента, и следовательно, — структура адсорбционного слоя. Мы полагаем, что дальнейшее развитие теории адсорбции невозможно без учета тех изменении в структуре растворов, которые происходят по мере повышения их концентрации.
Для проблемы поверхностных явлении в полимерах, и в частности для решения вопросов, связанных с адгезией, необходимо также исследование условий адсорбционного взаимодействия полимерных молекул с поверхностями в очень концентрированных системах или в отсутствие растворителя.
Из изложенного следует, что в области теории адсорбции имеется много нерешенных проблем, имеющих существенное значение для правильного понимания механизма процесса. Нельзя, однако, думать, что все нерешенные проблемы адсорбции относятся к области теории. Развитие теория задерживается еще и потому, что многие вопросы недостаточно исследованы экспериментально. К таким вопросам можно отнести экспериментальное исследование влияния на адсорбцию гибкости полимерной цепи в условиях, когда энергия взаимодействия цепи с поверхностью сохраняется постоянной. Нет прямых определений энергий адсорбции полимеров на твердых поверхностях, мало исследовано влияние полидисперсности на адсорбцию. В литературе отсутствуют работы по адсорбции блок- и привитых полимеров, которые могли бы дать существенные сведения об условиях адсорбционного взаимодействия, не рассмотрены проблемы адсорбции кристаллизующихся полимеров, олигомеров и т. п. Отсутствуют прямые экспериментальные данные о структуре адсорбционных слоев. Решение вопросов адсорбции имеет значение не только для теории, но и для практики. Собственно говоря, сама проблема выдвинута потребностями практики.
ЛИТЕРАТУРА
1. Ю. С. Л и п а т о в.-Вестy. АН УССР. 1970. 9. 38.
2.Ю.С. Липато в.— VI Юбилейная Всесоюзная конференция по коллоидной химии. Тезисы докладов. Изд. Воронежского ун-та. 1968, 54
3. Б.В. Дерягин. Н.А. Кротова. Адгсзия. Изд во АН СССР. М… 1949.
4. Н. А.Кротова. О склеивании и прилипания Изд-во АН СССР. М., 1960.
5. С.С. Воюцкий. Адгезия и аутогезия полимеров. Ростехиздат. М… 1963.
6. Адгезия. ИЛ. М. 1954.
7. П И. Москвитин. Склеивание полимеров «Лесная промьшленность». М… 1968.
8. А.А. Берлин, В.Е.Басян. Основы адгезии полимеров. «Химия». М… 1969.
9. Я.О.Бикерман.- Высокомолек. соед… 1968. A 10, 1974
10. С.С.Воюцкий, Б.В.Дерягин.- Коллоид, ж… 1965, 27, 624
11. Ю. С Липатов. Физика химия наполненных полимеров. «Наукова думка» К. 1967.
12. Б.А.Киселев Стеклопластики Госхимиздат. M… 1961.
13. Г.Л. Андреевская. Высокопрочные ориентированные стеклопластики, 1966
14. В.Б. Тихомиров. Физико-химические основы получения нетканых материалов, 1969.
15. Ю.С.Липатов, В. П. Максимова. Л.М.Сepreeва.- Высокомол. соед., 1960, 2, 596.
16. Ю. С. Липатов. Н Г. Перышкина, Л. М. Сергеев а.— Высокомол. соед., 1962. 4, 596.
17. Ю. С. Липатов, Л. М. Сергeева. — В кн.: Ионообмен и сорбция из растворов. Изд-во АН БССР. Минск. 1963. 63.
18. Ю.С.Липатов, Т. Э. Геллер.- Высокомолек. соед., 1966. 8, 582.
19. Ю.С.Липатов, Т. Э. Гелле р.- Высокомолек. соед., 1967. 9 А. 241.
20. Т. Э. Липатова. Я.С.Скорынина, Ю.С.Липатов Адгезия полимеров. АН СССР, М… 1963. 117
21. В.А.Ккаргин, Ю.С. Липатов.— Ж физ. химии, 1958. 32. 326
22. Ю.С.Липатов.- ДАН СССР. 1962. 143. 1142.
23. А.П.Кvксин, Л. М. Сергсева. Ю. С. Липатов, Л.И.Безрук.— Высокомол. соедин. 1970. А 12. 2332.
24. С. А. Шреинер. П. И. Зубов — Коллоид. ж… 1960. 22, 727.
25. П II. Зубов. А. Г.Санжаровский.— Лакокрас матер… 1963. 2. 48.
26. М. Азори, Н. А. Платэ и др.-Высокомолек. соединения. 1966, 8 759
27. В.А.Каргин. Т. И. Соколова, И. И. Курбанова. — ДАН СССР. 162, 1095. 1965.
28. В.А.Каргин. В.А.Кабанов.- ЖВХО. 1964. 9. 6021.
29. Л. М. Сергеева. Ю.С Липатов. В кн. «Синтез и физико-химия полиуретнов», «Наукова думка». К. 1967. 131.
30. Е. Б. Тростянская — В кн.: Наполнители полимерных материалов. Изд. Дом научно-техн. пропаганды им. Дзержинского. М., 1969. 3.
31. В. А. Каргин. Т. И. Соголова. И.Я. Рапопорт-Молодцова — Высокомол. соедин. 1964. 4, 2090.
32. Ю.М. Малинский и др.— Высоком. соед… 1968. А 10. 786.
33. Ю.М. Малинский и др.- ДАН СССР. 1965. 160. 1123.
34. Ю. С. Липатов, С.С.Крафчик. Ю.Ю. Керча.- ДАН УССР 1971.2. 143.
35. Ф. Г. Фабуляк. Ю. С. Липатов.- Высокомол. соед. 1970. Б 12. 871.